Recombinación homóloga
A recombinación homóloga é o tipo máis coñecido e común de recombinación xenética que consiste no intercambio de secuencias de nucleótidos entre dúas moléculas similares ou idénticas de ADN. A recombinación homóloga ocorre durante a meiose (tipo de división celular na que, nos animais, se forman os gametos) e na reparación do ADN e no intercambio de xenes entre microorganismos.

Na recombinación homóloga meiótica os cromosomas homólogos (do mesmo par cromosómico) intercambian fragmentos, orixinando novas combinacións de secuencias de ADN. As novas combinacións de ADN supoñen a introdución de variabilidade xenética na descendencia, o cal permitirá que as poboacións se adapten durante o decurso da evolución.[1] As células utilizan tamén moi estendidamente a recombinación homóloga para repararen con precisión as roturas que afectan ás dúas cadeas do ADN, denominadas roturas de dobre cadea. A recombinación homóloga utilízase tamén na transferencia horizontal de xenes para intercambiar material xenético entre distintas cepas de bacterias e virus.
Aínda que a recombinación homóloga varía moito entre diferentes organismos e tipos celulares, a maioría das formas implican os mesmos pasos básicos. Despois de producirse unha rotura de dobre cadea no ADN, córtanse e sepáranse as seccións de ADN situadas arredor dos extremos 5' do punto de rotura, proceso chamado resección. No paso seguinte chamado invasión da febra, un extremo 3' colgante da molécula rota de ADN "invade" despois unha molécula similar ou idéntica de ADN que non está rota. Despois da invasión da febra, as dúas moléculas de ADN quedan conectadas por dúas estruturas con forma de cruz chamadas unións de Holliday. Dependendo de como os encimas cortan estas dúas unións, o tipo de recombinación homóloga que ocorre na meiose poderá dar lugar ou ben a un sobrecruzamento cromosómico ou non haber sobrecruzamento. A recombinación homóloga que ocorre durante a reparación do ADN tende a orixinar produtos sen sobrecruzamento, o que restaura as moléculas de ADN danadas tal como estaban antes da rotura de dobre cadea.
A recombinación homóloga é un proceso conservado nos seres vivos, que se pode atopar nos tres dominios da vida e nos virus, polo que é un mecanismo case universal. O descubrimento en protistas de xenes para a recombinación homóloga interpretouse como unha evidencia de que a meiose xurdiu moi cedo na evolución dos eucariotas. Como a súa disfunción está fortemente asociada cun incremento da susceptibilidade a varios tipos de cancro, as proteínas que facilitan a recombinación homóloga están sendo investigadas activamente. A recombinación homóloga utilízase tamén no chamado gene targeting, unha técnica para introducir cambios xenéticos nos organismos. Polo desenvolvemento desta técnica, os investigadores Mario Capecchi, Martin Evans e Oliver Smithies foron galardoados co Premio Nobel de Medicina e Fisioloxía de 2007.
Historia
editar
A comezos da década de 1900, William Bateson e Reginald Punnett encontraron unha excepción nun dos principios da herdanza que foran orixinalmente descritos por Gregor Mendel na década de 1860. A noción de Mendel era que os trazos xenéticos se distribuían independentemente cando pasaban de pais a fillos; por exemplo, a cor dos ollos dun gato e a súa lonxitude do rabo herdábanse independentemente unha da outra. Pero Bateson e Punnett atoparon que certos xenes asociados con trazos físicos poden herdarse conxuntamente, é dicir, que están ligados xeneticamente.[2][3] En 1911, Thomas Hunt Morgan, despois de observar que os trazos ligados en certas ocasións tamén se podían herdar independentemente, suxeriu que poderían ocorrer "sobrecruzamentos" entre os xenes ligados,[4] nos cales un dos xenes ligados pasaba fisicamente a un cromosoma diferente. Dúas décadas despois, Barbara McClintock e Harriet Creighton demostraron que o sobrecruzamento cromosómico ten lugar durante a meiose,[5][6] (o proceso de división celular mediante o cal se forman os espermatozoides e óvulos). No mesmo ano en que se fixo o descubrimento de McClintock, Curt Stern demostrou que o sobrecruzamento, máis tarde chamado "recombinación", podía tamén ocorrer en células somáticas como os glóbulos brancos do sangue e as células da pel, que se dividen por mitose.[5][7]
En 1947, o microbiólogo Joshua Lederberg demostrou que as bacterias, que se pensaba que só se reproducían asexualmente por fisión binaria, podían tamén realizar unha recombinación xenética similar á da reprodución sexual. O seu traballo realizouse coa bacteria E. coli, que se converteu nun organismo modelo para estudos xenéticos,[8] e contribuíu a que Lederberg gañase o premio Nobel de Medicina de 1958.[9] En 1964 Robin Holliday, baseándose en estudos en fungos, propuxo un modelo para a recombinación na meiose no que incluíu detalles fundamentais sobre o posible funcionamento do proceso, como o intercambio de material entre os cromosomas por medio das unións ou estruturas de Holliday.[10] En 1983, Jack Szostak e colegas presentaron un modelo agora chamado vía DSBR, que podía explicar observacións non explicadas polo modelo de Holliday.[10][11] Durante a seguinte década, os experimentos en Drosophila, lévedos e células de mamíferos utilizáronse para presentar outros modelos de recombinación homóloga, chamados vías SDSA, que non sempre dependen das unións de Holliday.[10]
En eucariotas
editarA recombinación homóloga é esencial na división celular dos eucariotas (plantas, animais, fungos e protistas). Nas células que se dividen por mitose, a recombinación homóloga repara roturas de dobre cadea no ADN causadas por radiacións ionizantes ou substancias químicas que danan o ADN.[12] Se estas roturas de dobre cadea non se reparasen poderían causar rearranxos a grande escala nos cromosomas nas células somáticas,[13] o cal podería orixinar cancro.[14]
Ademais de reparar o ADN, a recombinación homóloga contribúe tamén a producir diversidade xenética cando as células se dividen na meiose para orixinar os gametos nos animais, pole e ovocélulas nas plantas, e esporas en certas plantas e fungos. A diversidade créase por recombinación por medio do sobrecruzamento cromosómico, na cal se intercambian rexións similares pero non idénticas do ADN entre cromosomas homólogos.[15][16] Isto crea novas combinacións de xenes que poden ser beneficiosas, e poden darlle á descendencia unha vantaxe evolutiva.[17] O sobrecruzamento cromosómico empeza cando unha proteína chamada Spo11 produce unha rotura de dobre cadea nun punto determinado do ADN.[18] Estes sitios non están localizados aleatoriamente nos cromosomas, senón que xeralmente se encontran en rexións promotoras interxénicas e preferentemente en zonas ricas en GC.[19] Estes sitios onde se producen as roturas de dobre cadea aparecen a miúdo en puntos quentes de recombinación, que son rexións dos cromosomas dunha lonxitude de 1.000–2.000 pares de bases, que presentan unha alta frecuencia de recombinación. A ausencia de puntos quentes de recombinación entre dous xenes do mesmo cromosoma a miúdo supón que eses xenes os herdarán as futuras xeracións en igual proporción. Isto representa que hai un ligamento entre os dous xenes maior do que se esperaría por distribución independente mendeliana durante a meiose.[20]
A recombinación no ciclo celular
editar
As roturas de dobre cadea poden ser reparadas por medio da recombinación homóloga ou pola unión de extremos non homólogos (NHEJ, do inglés non-homologous end joining). A unión de extremos non homólogos é un mecanismo de reparación do ADN que, a diferenza da recombinación homóloga, non necesita que exista unha longa secuencia homóloga para guiar a reparación. A fase do ciclo celular na que se atopa a célula é a que determina en grande medida se se utilizará a recombinación homóloga ou ben a unión de extremos non homólogos para reparar as roturas de dobre cadea. A recombinación homóloga repara o ADN antes de que a célula entre na mitose (fase M). Ten lugar durante e pouco despois da replicación do ADN, durante as fases S e G2 do ciclo celular, cando as cromátides irmás son máis accesibles.[21] Comparados cos cromosomas homólogos, que son similares a outros cromosomas pero que a miúdo teñen alelos diferentes, as cromátides irmás son un molde ideal para a recombinación homóloga porque son copias idénticas dun determinado cromosoma. A diferenza da recombinación homóloga, a unión de extremos non homólogos ocorre predominantemente na fase G1 do ciclo celular, cando a célula está a crecer pero non está aínda preparada para dividirse. Ocorre tamén menos frecuentemente despois da fase G1, pero mantén polo menos algunha actividade en todas as fases do ciclo celular. Os mecanismos que regulan a recombinación homóloga e a unión de extremos non homólogos ao longo do ciclo celular varían moito entre as especies.[22]
As quinases dependentes de ciclina (CDKs), que modifican a actividade doutras proteínas ao engadirlles grupos fosfato (fosforilándoas), son reguladores importantes na recombinación eucariótica.[22] Cando comeza a replicación do ADN nos lévedos, a quinase dependente de ciclina Cdc28 inicia a recombinación homóloga fosforilando á proteína Sae2.[23] Despois de ser activada pola adición do fosfato, Sae2 usa a súa actividade de endonuclease para facer un corte cerca dunha rotura de dobre cadea do ADN. Isto permite que se una ao ADN un complexo proteico formado por tres compoñentes chamado complexo MRX, e comece unha serie de reaccións dirixidas por proteínas que realizan o intercambio de material entre dúas moléculas de ADN.[24]
Modelos
editarDous modelos primarios para explicar como a recombinación homóloga repara as roturas de dobre cadea do ADN son a vía de reparación de roturas de dobre cadea (DSBR, do inglés double-strand break repair, ás veces tamén chamada modelo de unión de Holliday dobre ou DHJ) e a vía de annealing de febra dependente de síntese (SDSA, do inglés synthesis-dependent strand annealing).[25] Ambas as vías son similares nos seus pasos iniciais. Despois de que se produce unha rotura de dobre cadea, o complexo MRX (en humanos chamado complexo MRN) únese ao ADN en cada lado da rotura. Seguidamente, realízase un novo corte en dúas fases, no cal se corta o ADN que está arredor dos extremos 5' da rotura. No primeiro paso deste corte, o complexo MRX recruta a proteína Sae2. As dúas proteínas despois fan o corte (resección) dos extremos 5' a cada lado da rotura para crear un curto tramo 3' monocatenario que sobresae. No segundo paso, a helicase Sgs1 e a Exo1 e a Dna2 continúan o corte 5'→3'. A helicase Sgs1 desenrola a dobre hélice do ADN bicatenario, mentres que a actividade nuclease de Exo1 e Dna2 corta o tramo de ADN monocatenario producido pola Sgs1.[23]

A proteína de replicación A (RPA), que ten unha alta afinidade polo ADN monocatenario, únese despois aos tramos saíntes 3' colgantes.[26] Coa axuda doutras proteínas que median no proceso, a proteína Rad51 (e Dmc1, na meiose) forma despois un filamento de ácido nucleico e proteína no ADN bicatenario cuberto coa RPA. Este filamento de nucleoproteína empeza a procurar secuencias de ADN similares aos do tramo saínte 3'. Unha vez que encontra unha, o filamento de nucleoproteína monocatenario móvese (invade) ao ADN dúplex receptor idéntico ou similar nun proceso que se chama invasión da febra. Nas células que se dividen por mitose, o ADN dúplex receptor é xeralmente unha cromátide irmá, que é idéntica á molécula de ADN danada e proporciona un molde para a reparación. Porén, na meiose, o ADN receptor adoita pertencer a un cromosoma homólogo similar pero non necesariamente idéntico.[25] Fórmase un bucle de desprazamento (bucle D) durante a invasión da febra entre o tramo saínte 3' e o cromosoma homólogo. Despois da invasión da febra, unha ADN polimerase alonga o extremo da febra invasora 3' sintetizando novo ADN. Isto cambia a forma do bucle D, que pasa a ser unha estrutura con forma de cruz chamada unión de Holliday (Holliday junction). Seguidamente, ten lugar máis síntese de ADN na febra invadida (é dicir, un dos tramos saíntes 3' orixinais), restaurando a febra no cromosoma homólogo que fora desprazada durante a invasión da febra.[25]
Vía DSBR
editarDespois das fases de resección, invasión da febra e síntese de ADN, as vías DSBR e SDSA son distintas.[25] Un fenómeno exclusivo da vía DSBR é que o segundo tramo saínte 3' (que non estaba implicado na invasión da febra) tamén forma unha unión de Holliday co cromosoma homólogo. As unións de Holliday dobres son despois convertidas en produtos de recombinación por endonucleases que crean amosegas no ADN (nicking endonucleases), un tipo de endonuclease de restrición, que corta só unha das cadeas do ADN. A vía DSBR dá lugar xeralmente a sobrecruzamento, aínda que pode ás veces orixinar produtos sen sobrecruzamento; a capacidade dunha molécula de ADN rota de recoller secuencias de loci doantes separados demostrouse en lévedos de xemación en mitose utilizando plásmidos ou a indución de eventos cromosómicos por endonuclease.[27][28] Debido a esta tendencia que ten a vía DSBR ao sobrecruzamento cromosómico, é un modelo probable do mecanismo da recombinación homóloga na meiose.[15]
Que a recombinación pola vía DSBR dea lugar ou non a sobrecruzamento cromosómico está determinado pola maneira en que se corta ou "resolve" a unión de Holliday dobre. Prodúcese o sobrecruzamento cromosómico se unha unión de Holliday se corta na febra que cruza e a outra unión de Holliday se corta na febra que non cruza (na Figura 4, ao longo das puntas de frecha púrpuras nunha unión de Holliday e ao longo das puntas de frecha laranxas verticais na outra). Alternativamente, se as dúas unións de Holliday se cortan nas febras que cruzan (ao longo das cabezas de frecha púrpuras de ambas as unións de Holliday da Figura 4), entón vanse orixinar cromosomas sen sobrecruzamento.[29]
Vía SDSA
editarA recombinación homóloga pola vía SDSA ocorre en células que se dividen por mitose e orixina produtos sen sobrecruzamento. Neste modelo, a febra 3' invasora é producida sobre o ADN dúplex receptor por unha ADN polimerase, e libérase a medida que a unión de Holliday entre as moléculas de ADN doante e receptora escorrega nun proceso chamado migración da rama (branch migration). O extremo 3' de nova síntese da febra invasora pode despois aparearse (anneal) co outro tramo saínte 3' no cromosoma danado por medio dun apareamento de bases complementarias. Despois do aliñamento das febras, pode quedar por veces unha pequena solapa de ADN. Todas estas posibles solapas son eliminadas, e a vía SDSA remata co reselado do ADN, tamén chamado ligazón, de calquera intervalo de cadea simple que puidese quedar.[30]
Vía SSA
editar
Outra vía de recombinación homóloga é a vía SSA (do inglés single-strand annealing). A vía SSA da recombinación homóloga repara roturas de dobre cadea entre dúas secuencias repetidas. A vía SSA non require unha molécula de ADN idéntica ou similar separada, como as vías DSBR ou SDSA. O único que require a vía SSA é un ADN dúplex, e utiliza as secuencias repetidas como as secuencias idénticas que necesita a recombinación homóloga para a reparación. A vía é relativamente simple no seu concepto: despois de que se cortan de novo as dúas cadeas do mesmo ADN dúplex arredor do sitio onde se produciu a rotura de dobre cadea, os dous tramos saíntes 3' resultantes alíñanse e aparéanse entre si, volvendo a formar un dúplex continuo de ADN.[30][31]
A medida que se corta de novo o ADN arredor da rotura de dobre cadea, os tramos saíntes 3' producidos son cubertos pola proteína RPA, que impide que os tramos saíntes 3' se peguen entre si.[32] Despois, unha proteína chamada Rad52 únese a cada unha das secuencias repetidas en cada lado da rotura, e alíñaas para permitir que as dúas secuencias repetidas complementarias se apareen.[32] Despois de que se completou o apareamento, as solapas non homólogas sobrantes dos tramos saíntes 3' córtanse e elimínanse por un conxunto de nucleases denominadas Rad1/Rad10, que as proteínas Saw1 e Slx4 traen ata as solapas.[32][33] Os intervalos ocos que queden son enchidos con ADN de nova síntese, e a ligación restaura o ADN dúplex que volve a ser de dúas febras continuas.[34] A secuencia de ADN entre as repeticións pérdese sempre, igual que unha das dúas repeticións. A vía SSA considérase mutaxénica xa que orixina as mencionadas delecións de material xenético.[30]
Vía BIR
editarDurante a replicación do ADN, poden ás veces atoparse roturas de dobre cadea en forquitas de replicación a medida que a ADN helicase desenrola a cadea molde. Estes defectos son reparados na vía da replicación inducida por rotura (BIR, do inglés break-induced replication) da recombinación homóloga. Os mecanismos moleculares precisos da vía BIR non están aínda claros. Propuxéronse tres mecanismos que teñen a invasión de febra como paso inicial, pero difiren en como explican a migración do bucle D e as fases posteriores da recombinación.[35]
A vía BIR pode tamén axudar a manter a lonxitude dos telómeros (rexións do ADN no extremo de cromosomas eucarióticos) en ausencia do encima telomerase ou cooperando con el. Se non hai copias funcionais de telomerase, os telómeros acórtanse en cada ciclo de mitose, o cal finalmente bloquea a división celular e leva á senescencia celular. Nos lévedos nos que a telomerase foi inactivada por mutacións, observáronse dous tipos de células "superviventes" que evitaban a chegada á senescencia durante máis tempo do agardado elongando os seus telómeros por medio das vías BIR.[35]
O mantemento da lonxitude dos telómeros é fundamental para a inmortalización de células, que é unha característica do cancro. A maioría das células de cancros manteñen a lonxitude dos telómeros regulando á alza o encima telomerase. Non obstante, en varios tipos de cancro humano, unha vía de tipo BIR contribúe a manter algúns tumores actuando como un mecanismo alternativo de mantemento dos telómeros.[36] Este feito levou aos científicos a investigar se tales mecanismos de mantemento dos telómeros baseados na recombinación poderían desbaratar os efectos de drogas anticancerosas como os inhibidores da telomerase.[37]
En bacterias
editarA recombinación homóloga é un proceso de reparación do ADN de grande importancia en bacterias. Tamén é importante para producir diversidade xenética en poboacións bacterianas, aínda que o proceso é substancialmente diferente da recombinación meiótica, que produce diversidade nos organismos eucarióticos. Onde foi máis estudada e se comprende mellor a recombinación homóloga bacteriana é en Escherichia coli.[38] As roturas de dobre cadea en bacterias son reparadas pola vía RecBCD de recombinación homóloga. As roturas que ocorren só nunha das dúas febras do ADN, coñecidas como ocos (gaps) de cadea simple, crese que se reparan pola vía RecF (véxase máis abaixo).[39] As vías RecBCD e RecF inclúen series de reaccións chamadas migración da rama (na cal unha febra simple de ADN se intercambia con outra entre dúas moléculas entrecruzadas de ADN dúplex) e resolución (na cal estas dúas moléculas entrecruzadas de ADN son cortadas e separadas e son restauradas no seu estado normal de dobre cadea).
Vía RecBCD
editar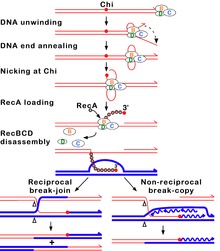

A vía RecBCD é a principal vía de recombinación utilizada para reparar roturas de dobre enlace no ADN.[42][43] Estas roturas de dobre enlace poden ser causadas pola luz ultravioleta e outras radiacións ionizantes, ou por mutáxenos químicos. As roturas de dobre enlace poden orixinarse tamén pola replicación do ADN nunha zona onde hai unha amosega nunha das cadeas do ADN ou nun oco (gap) onde faltan nucleótidos nunha das cadeas. Esas situacións causan o que se denomina forquita de replicación colapsada e son reparadas por varias vías de recombinación homóloga entre as que está a vía RecBCD.[44]
Nesta vía, un complexo encimático de tres subunidades chamado RecBCD inicia a recombinación uníndose a un extremo romo ou case romo dunha rotura de dobre cadea do ADN. Unha vez que RecBCD se une ao extremo do ADN, as subunidades RecB e RecD empezan a desenrolar o ADN dúplex por medio de actividade helicase. A subunidade RecB tamén ten un dominio nuclease, que corta a febra simple de ADN que se forma polo proceso de desenrolamento. Este desenrolamento continúa ata que RecBCD encontra un secuencia de nucleótidos específica (5'-GCTGGTGG-3') chamada sitio Chi (crossover hotspot instigator, instigador de punto quente de sobrecruzamento).[43]
Despois de atoparse co sitio Chi, a actividade do encima RecBCD cambia drasticamente.[42][45][46] O desenrolamento do ADN para durante uns segundos e despois volve a comezar a aproximadamente a metade da velocidade inicial. No desenrolamento interveñen as helicases RecB (máis lenta) e RecD (máis rápida). Que o desenrolamento vaia agora á metade da velocidade débese probablemente a que é a helicase máis lenta RecB a que desenrola o ADN despois do sitio Chi, en vez de facelo a helicase máis rápida RecD, que só desenrolaría o ADN antes de Chi.[47][48] O rcoñecemento do sitio Chi tamén cambia o estado do encima RecBCD para que corte a febra de ADN que ten o sitio Chi e empece a cargar moitas proteínas RecA no ADN monocatenario co extremo 3' novo xerado. O filamento nucleoproteico cuberto por RecA resultante busca despois secuencias de ADN similares no cromosoma homólogo. O proceso de busca induce o estiramento do ADN dúplex, o cal mellora o recoñecemento da homoloxía (un mecanismo chamado corrección de probas conformacional [49][50] [51]). Unha vez encontrada unha secuencia axeitada, o filamento nucleoproteico monocatenario móvese ao ADN dúplex receptor homólogo nun proceso chamado invasión da febra.[52] O tramo saínte 3' causa que unha das febras do ADN dúplex receptor se desprace e se forme o bucle D. Se se corta o bucle D, outro intercambio de febras orixina unha estrutura en forma de cruz chamada unión de Holliday.[43] A resolución da unión de Holliday por algunhas combinacións de RuvABC ou RecG pode producir dúas moléculas de ADN recombinantes con tipos xenéticos recíprocos, se as dúas moléculas de ADN que interaccionsn difiren xeneticamente. Alternativamente, o extremo 3' invasor preto de Chi pode cebar a síntese de ADN e formar unha forquita de replicación. Este tipo de resolución produce só un tipo de recombinante (non reciproco).
Vía RecF
editarAs bacterias parecen usar a vía RecF da recombinación homóloga para reparar ocos nunha soa cadea do ADN (onde faltan nucleótidos). Cando mutacións inactivan a vía RecBCD e outras mutacións adicionais inactivan as nucleases SbcCD e ExoI, a vía RecF pode tamén reparar as roturas de dobre cadea do ADN.[53] Na vía RecF a helicase RecQ desenrola o ADN e a nuclease RecJ degrada a febra que ten o extremo 5’, deixando a febra co extremo 3' intacta. A proteína RecA únese a esta febra e ou ben é axudada polas proteínas RecF, RecO, e RecR ou estabilizada por elas. O filamento nucleoproteico RecA despois busca un ADN homólogo e intercambia o seu posto coa febra idéntica ou case idéntica no ADN homólogo.
Aínda que as proteínas e os mecanismos específicos implicados nas súas fases iniciais son diferentes, as dúas vías son similares en que ambas requiren un ADN monocatenario cun extremo 3’ e a proteína RecA para facer a invasión da febra. As vías son tamén similares nas súas fases de migración da rama, nas cales a unión de Holliday escorrega nunha dirección, e na de resolución, na que as unións de Holliday son clivadas e retiradas por encimas.[54][55] O tipo non recíproco alternativo de resolución pode tamén ocorrer por ambas as vías.
Migración da rama
editarInmediatamente despois da invasión da febra, a unión de Holliday móvese ao longo do ADN ligado durante o proceso de migración da rama. É neste movemento da unión de Holliday cando se intercambian os apareamentos de bases entre os dous ADN dúplex homólogos. Para catalizar a migración da rama, a proteína RuvA recoñece e despois únese á unión de Holliday e recruta a proteína RuvB para formar o complexo RuvAB. Cárganse dous conxuntos de proteínas RuvB (cada un formará unha ATPase con forma de anel), en lados opostos da unión de Holliday, onde actúan como bombas xemelgas que proporcionan a forza para que se produza a migración da rama. Entre os dous aneis de RuvB, ensámblanse dous conxuntos da proteína RuvA no centro da unión de Holliday, de modo que o ADN na unión queda no medio dos dous conxuntos de RuvA. As febras de ambos os dúplex de ADN (o "doante" e o "receptor") son desenroladas sobre a superficie de RuvA a medida que son guiadas pola proteína desde un dos dúplex ao outro.[56][57]
Resolución
editarNa fase de resolución da recombinación, córtanse todas as unións de Holliday que se forman polo proceso de invasión da febra, polo que se volve a recuperar o estado de dúas moléculas separadas de ADN. Esta clivaxe (corte) faina o complexo RuvAB interaccionando con RuvC, que forman xuntos o complexo RuvABC. RuvC é unha endonuclease que corta a secuencia dexenerada 5'-(A/T)TT(G/C)-3'. esta secuencia encóntrase frecuentemente no ADN, aproximadamente unha vez cada 64 nucleótidos.[57] Antes de cortar, probablemente RuvC accede á unión de Holliday desprazando un dos dous tetrámeros de RuvA que cubrían alí o ADN.[56] A recombinación orixina produtos de "empalme" ("splice") ou de "parche" ("patch"), dependendo de como RuvC cliva a unión de Holliday.[57] Os produtos de empalme son produtos de sobrecruzamento, nos cales hai un rearranxo de material xenético arredor do sitio de recombinación. Os produtos de parche son produtos sen sobrecruzamento nos cales non hai ese rearranxo e só hai un "parche" de ADN híbrido no produto recombinante.[58]
Facilitación da transferencia xenética
editarA recombinación homóloga é un importante método para integrar ADN doante no xenoma dun organismo receptor na transferencia horizontal de xenes, que é o proceso polo cal un organismo incorpora ADN alleo procedente doutro organismo sen ser un descendente dese organismo. A recombinación homóloga require que o ADN entrante sexa moi similar ao do xenoma receptor, polo que a transferencia horizontal de xenes está normalmente limitada a bacterias similares.[59] Os estudos realizados en varias especies de bacterias estableceron que hai un decrecemento logarítmico-linear na frecuencia de recombinación cando se incrementan as diferenzas na secuencia entre o ADN do hóspede e o receptor.[60][61][62]
Na conxugación bacteriana, na cal o ADN se transfire dunha bacteria a outra por contacto directo entre as células, a recombinación homóloga axuda a integrar o ADN alleo no xenoma do hóspede por medio da vía RecBCD. O encima RecBCD promove a recombinación unha vez que o ADN, que entrou inicialmente na bacteria como monocatenario, se converte en ADN bicatenario durante a replicación.
A vía RecBCD é tamén esencial na fase final da transdución, que é un tipo de transferencia horizontal de xenes na cal o ADN se transfire dunha bacteria a outra por intermediación dun virus. A transdución prodúcese por exemplo porque ás veces se incorpora ADN bacteriano alleo á cápside dun virus bacteriófago no momento en que o ADN se está a empaquetar dentro dos novos bacteriófagos formados durante a replicación viral. Cando estes novos bacteriófagos infectan a outra bacteria, o ADN procedente da anterior bacteria hóspede do virus é inxectado na nova bacteria hóspede como ADN bicatenario. Despois, o encima RecBCD incorpora este ADN bicatenario no xenoma da nova bacteria hóspede.[43]
En virus
editarA recombinación homóloga dáse tamén en varios grupos de virus. En virus de ADN como os herpesvirus, a recombinación ocorre por medio dun mecanismo de rotura e reunión como en bacterias e eucariotas.[63] Hai tamén evidencias de recombinación nalgúns virus de ARN, especificamente os virus de ARN monocatenario de sentido positivo como os retrovirus, picornavirus, e coronavirus. Hai controversia sobre se a recombinación homóloga ocorre tamén en virus de ARN monocatenario de sentido negativo como o da gripe.[64]
Nos virus de ARN a recombinación homóloga pode ser precisa ou imprecisa. No tipo preciso de recombinación ARN-ARN, non hai diferenza entre as dúas secuencias parentais de ARN e a rexión de ARN de sobrecruzamento resultante. Debido a isto, é con frecuencia difícil determinar a localización dos eventos de sobrecruzamento entre as dúas secuencias recombinantes. Na recombinación homóloga de ARNs de tipo impreciso, a rexión de sobrecruzamento ten algunhas diferenzas con respecto ás secuencias do ARN parental, causadas por adición, deleción, ou outras modificacións de nucleótidos. O nivel de precisión no sobrecruzamento está controlado polo contexto da secuencia das dúas febras recombinantes de ARN: as secuencias ricas en adenina e uracilo fan diminuír a precisión do sobrecruzamento.[65]
A recombinación homóloga é importante para facilitar a evolución viral.[65][66] Por exemplo, se o xenoma de dous virus con diferentes mutacións desvantaxosas sofre recombinación, entón poden ter a capacidade de rexenerar un xenoma completamente funcional. Alternativamente, se dous virus similares infectaron á mesma célula hóspede, a recombinación homóloga pode permitir que eses dous virus intercambien xenes e así evolucionen variantes máis potentes deses virus.[66]
A recombinación homóloga é o mecanismo proposto para explicar a integración do virus de ADN herpesvirus humano 6 nos telómeros humanos.[67]
Efectos da disfunción
editarSe a recombinación homóloga non se realiza axeitadamente, os cromosomas se aliñarían incorrectamente con frecuencia na profase I da división celular meiótica. Isto fai que os cromosomas non poidan segregarse debidamente, fenómeno chamado non disxunción. Á súa vez, a non disxunción pode causar que os espermatozoides e óvulos teñan un exceso ou un defecto de cromosomas. A síndrome de Down, que é causada xeralmente por ter unha copia extra do cromosoma 21, é unha das moitas anormalidades resultantes destes fallos na recombinación homóloga meiótica.[57][68]
As deficiencias na recombinación homóloga están moi asociadas coa formación de tumores en humanos. Por exemplo, as síndromes de Bloom, de Werner e de Rothmund-Thomson, que están relacionadas co cancro, son causadas polo mal funcionamento de copias dos xenes da helicase RecQ implicados na regulación da recombinación homóloga, chamados, respectivamente, BLM, WRN e RECQ4.[69] Nas células dos pacientes que teñen a síndrome de Bloom, que carecen dunha copia funcional da proteína BLM, hai unha elevada frecuencia de recombinación homóloga.[70] Os resultados dalgúns experimentos realizados en ratos deficientes para a BLM suxiren que a mutación dá lugar a cancro por medio dunha perda de heterocigose causada polo incremento na recombinación homóloga.[71] Unha perda de heterocigose indica a perda dunha das dúas versións (alelos) dun xene. Se un dos alelos perdidos contribuía a suprimir tumores, como o xene da proteína do retinoblastoma por exemplo, entón a perda da heterocigose pode orixinar un cancro.[72]
Cando a frecuencia de recombinación homóloga diminúe, isto causa que haxa unha reparación ineficaz do ADN,[72] o cal pode tamén orixinar cancro.[73] O mesmo ocorre cos xenes BRCA1 e BRCA2, que son xenes supresores de tumores, cuxo mal funcionamento está asociado cun incremento considerable do risco de padecer cancro de mama e de ovario. As células que non teñen BRCA1 e BRCA2 presentan unha frecuencia diminuída de recombinación homóloga e un incremento da sensibilidade á radiación ionizante, o que suxire que o decrecemento da recombinación homóloga leva a un incremento da susceptibilidade ao cancro.[73] Como a única función coñecida de BRCA2 é axudar a iniciar a recombinación homóloga, especulouse sobre se un coñecemento máis detallado do papel de BRCA2 na recombinación homóloga podería ser a chave para entender mellor as causas dos cancros de mama e ovario.[73]
Conservación evolutiva
editar
Aínda que as vías da recombinación poden variar no seu mecanismo, a capacidade dos organismos de realizar a recombinación homóloga está conservada universalmente en todos os dominios da vida.[74] Baseándose na semellanza das súas secuencias de aminoácidos, poden encontrarse homólogos de varias proteínas implicadas na recombinación en varios dominios da vida, o que indica que evolucionaron hai moito tempo, e que desde entón diverxiron a partir de proteínas ancestrais comúns.[74] Un destes conxuntos de proteínas é a familia proteica RecA/Rad51, que inclúe a proteína RecA de bacterias, a Rad51 e Dmc1 de eucariotas e a RadA e RadB de arqueas. Estas proteínas exercen funcións fundamentais nas fases iniciais da recombinación homóloga nos organismos que as expresan. As proteínas da familia RecA/Rad51 comparten unha rexión conservada longa chamada dominio RecA/Rad51. Neste dominio proteico hai dous motivos de secuencia chamados motivos de Walker A e B. Os motivos de Walker A e B permiten que os membros da familia proteica RecA/Rad51 realicen a hidrólise do ATP,[74] que proporciona enerxía para que as proteínas dirixan as reaccións da recombinación homóloga.[75]
Os estudos que modelizan as relacións evolutivas entre as proteínas Rad51, Dmc1 e RadA indican que son monofiléticas, ou que comparten un antepasado molecular común.[74] Dentro desta familia proteica, Rad51 e Dmc1 agrúpanse xuntas nun clado separado de RadA. Unha das razóns para agrupar estas tres proteínas é que todas posúen un motivo hélice-xiro-hélice modificado situado preto do seu extremo N-terminal, que contribúe a que as proteínas se unan ao ADN.[74] Propúxose un evento de duplicación xénica antigo do xene eucariótico RecA e unha posterior mutación como a orixe probable dos modernos xenes RAD51 e DMC1 (escritos agora con maiúsculas seguindo a nomenclatura dos xenes).[74]
O descubrimento da proteína Dmc1 en varias especies do protozoo Giardia, un dos primeiros protistas que diverxiu como eucariota, suxire que a recombinación meiótica homóloga (e a propia meiose) apareceron moi cedo na evolución dos eucariotas.[76] Ademais das investigacións sobre Dmc1, os estudos sobre a proteína Spo11 forneceron tamén moita información sobre as orixes da recombinación meiótica.[77] A proteína Spo11 é unha topoisomerase de tipo II que inicia a recombinación homóloga na meiose creando roturas de dobre cadea en puntos concretos do ADN.[18] As árbores filoxenéticas elaboradas baseándose na secuencia de xenes similares a SPO11 en animais, fungos, plantas, protistas e arqueas levaron aos científicos a crer que a versión Spo11 actual dos eucariotas xurdiu no último antepasado común de eucariotas e arqueas.[77]
Aplicacións tecnolóxicas
editarGene targeting
editar
- Artigo principal: Gene targeting.
O proceso da recombinación homóloga utilízase en moitos métodos para introducir secuencias de ADN en organismos para crear ADN recombinante e organismos modificados xeneticamente.[78] Métodos deste tipo, chamados gene targeting, úsanse especialmente en lévedos de xemación e en ratos. O método do gene targeting en ratos knockout usa células nai embrionarias de ratos para introducir material xenético artificial (principalmente de interese terapéutico), que reprime o xene diana do rato que interesa polo principio da recombinación homóloga. O rato actúa como un modelo de traballo para comprender os efectos dun xene de mamífero específico. En recoñecemento do seu descubrimento de como se pode utilizar a recombinación homóloga para introducir modificacións xenéticas en ratos por medio de células nai embrionarias, Mario Capecchi, Martin Evans e Oliver Smithies foron galardoados co Premio Nobel de Medicina de 2007.[79]
Os avances nas tecnoloxías do gene targeting, que aproveitan os mecanismos da recombinación homóloga das células están levando ao desenvolvemento dunha nova onda de modelos de enfermidades humanas isoxénicos máis precisos. Estes modelos de células humanas obtidas por enxeñaría pénsase que reflicten de forma máis fiel a xenética das enfermidades humanas que os modelos de ratos que se usaban anteriormente. Isto débese en grande medida a que se introducen as mutacións que interesan nos xenes endóxenos, tal como estas aparecen nos pacientes reais, e porque están baseadas nos xenomas humanos e non nos do rato. Ademais, certas tecnoloxías permiten o knock-in dunha mutación determinada en lugar de só os knock-outs que se aplicaban coas vellas tecnoloxías de gene targeting.
Enxeñaría de proteínas
editarA enxeñaría de proteínas con recombinación homóloga trata de preparar proteínas quiméricas intercambiando fragmentos entre dous proteínas parentais. Estas técnicas aproveitan o feito de que a recombinación pode introducir un alto grao de diversidade de secuencia á vez que preserva a capacidade da proteína de pregarse na súa estrutura terciaria, ou a súa forma tridimensional.[80] Isto supón unha diferenza con respecto a outras técnicas de enxeñaría de proteínas, como a mutaxénese de punto aleatorio, na cal a probabilidade de manter a funcionalidade da proteína declina expoñencialmente ao incrementárense as substitucións de aminoácidos.[81] As proteínas quimeras producidas por técnicas de recombinación poden manter a súa capacidade de pregarse porque os fragmentos parentais intercambiados están conservados estrutural e evolutivamente. Estes "ladrillos" para a construción de proteínas recombinables conservan interaccións importantes estruturalmente como os puntos de contacto físico entre diferentes aminoácidos da estrutura da proteína. Poden usarse métodos computacionais como o SCHEMA e a análise acoplada estatística para identificar subunidades estruturais axeitadas para a recombinación.[82][83][84]
As técnicas que se basean na recombinación homóloga utilizáronse para producir por enxeñaría novas proteínas.[82] Nun estudo publicado en 2007, infórmase da creación de quimeras de dous encimas implicados na biosíntese de isoprenoides, que son un tipo moi diverso de compostos entre os que hai hormonas, pigmentos visuais (carotenoides) e certas feromonas. As proteínas quiméricas adquiriron a capacidade de catalizar unha reacción esencial na biosíntese de isoprenoides, que é unha das vías biosintéticas máis diversas que hai na natureza, que estaba ausente nas proteínas parentais.[85] A enxeñaría de proteínas por medio de recombinación produciu tamén encimas quiméricos con novas funcións en proteínas da familia do citocromo P450,[86] que nos humanos está implicada na detoxificación de compostos alleos ao organismo como fármacos, aditivos alimentarios e conservantes.[87]
Terapia do cancro
editarAs células cancerosas con mutacións BRCA presentan deficiencias na recombinación homóloga, e desenvolvéronse fármacos que aproveitan estas deficiencias, que foron usados con éxito en ensaios clínicos.[88][89] O inhibidor de PARP1 Olaparib, contrae ou detén o crecemento dos tumores de mama, ovario e próstata causados polas mutacións dos xenes BRCA1 ou BRCA2, que son necesarios para a recombinación homóloga. Cando están ausentes os xenes BRCA1 ou BRCA2, outros tipos de mecanismos de reparación do ADN deben compensar a deficiencia na recombinación homóloga, como pode ser a reparación por escisión de bases (BER) para forquitas de replicación que quedaron paradas ou a unión de extremos non homólogos (NHEJ) para as roturas de dobre cadea.[88] Ao inhibir a BER nunha célula deficiencte na recombinación homóloga, Olaparib aplica o concepto de letalidade sintética para actuar especificamente sobre as células cancerosas. Aínda que os inhibidores do encima PARP1 representan un novo enfoque para a terapia do cancro, os investigadores advirten que poden ser insuficientes para a tratar cancros metastáticos en fases terminais.[88] As células cancerosas poden facerse resistentes ao inhibidor de PARP1 se sofren delecións de mutacións en BRCA2, o que evita a letalidade sintética dos fármacos ao recuperarse a capacidade das células cancerosas de reparar o ADN por medio da recombinación homóloga.[90]
Notas
editar- ↑ Alberts, B; et al. (2002). "Chapter 5: DNA Replication, Repair, and Recombination". Molecular Biology of the Cell (4th ed.). New York: Garland Science. p. 845. ISBN 0-8153-3218-1. OCLC 145080076.
- ↑ Bateson, P (August 2002). "William Bateson: a biologist ahead of his time" (PDF). Journal of Genetics 81 (2): 49–58. PMID 12532036. doi:10.1007/BF02715900.
- ↑ "Reginald Crundall Punnett". NAHSTE, University of Edinburgh. Consultado o 3 July 2010.
- ↑ Lobo, I; Shaw, K (2008). "Thomas Hunt Morgan, genetic recombination, and gene mapping". Nature Education 1 (1).
- ↑ 5,0 5,1 Coe, E; Kass, LB (10 May 2005). "Proof of physical exchange of genes on the chromosomes". PNAS 102 (19): 6641–6646. PMC 1100733. PMID 15867161. doi:10.1073/pnas.0407340102.
- ↑ Creighton, HB; Barbara, B (August 1931). "A correlation of cytological and genetical crossing-over in Zea Mays". PNAS 17 (8): 492–497. PMC 1076098. PMID 16587654. doi:10.1073/pnas.17.8.492.
- ↑ Stern, C (1931). "Zytologisch-genetische untersuchungen alsbeweise fur die Morgansche theorie des faktoraustauschs". Biol. Zentbl. 51: 547–587.
- ↑ Fee, E; et al. "The development of bacterial genetics". US National Library of Medicine. Consultado o 3 July 2010.
- ↑ "The Nobel Prize in Physiology or Medicine 1958". Nobelprize.org. Consultado o 3 July 2010.
- ↑ 10,0 10,1 10,2 Haber, JE; Ira, G; Malkova, A; Sugaware, N (29 Jan 2004). "Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday's model". Philos Trans R Soc Lond B Biol Sci 359 (1441): 79–86. PMC 1693306. PMID 15065659. doi:10.1098/rstb.2003.1367.
- ↑ Szostak, JW; Orr-Weaver, FW; Rothstein, RJ; Stahl, FW (May 1983). "The double-strand-break repair model for recombination". Cell 33 (1): 25–35. PMID 6380756. doi:10.1016/0092-8674(83)90331-8.
- ↑ Lodish H; et al. (2000). "12.5: Recombination between Homologous DNA Sites: Double-Strand Breaks in DNA Initiate Recombination". Molecular Cell Biology (4th ed.). W. H. Freeman and Company. ISBN 0-7167-3136-3.
- ↑ Griffiths, AJF; et al. (1999). "8: Chromosome Mutations: Chromosomal Rearrangements". Modern Genetic Analysis. W. H. Freeman and Company. ISBN 0-7167-3118-5.
- ↑ Kumma, KK; Jackson, SP (2001). "DNA double-strand breaks: signaling, repair and the cancer connection". Nature Genetics 27 (3): 247–254. PMID 11242102. doi:10.1038/85798.
- ↑ 15,0 15,1 Nelson, DL; Cox, MM (2005). Principles of Biochemistry (4th ed.). Freeman. pp. 980–981. ISBN 978-0-7167-4339-2.
- ↑ Marcon, E; Moens, PB (August 2005). "The evolution of meiosis: recruitment and modification of somatic DNA-repair proteins". BioEssays 27 (8): 795–808. PMID 16015600. doi:10.1002/bies.20264.
- ↑ Alberts B; et al. (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 305. ISBN 978-0-8153-4105-5.
- ↑ 18,0 18,1 Keeney, S; Giroux, CN; Kleckner, N (7 February 1997). "Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family". Cell 88 (3): 375–384. PMID 9039264. doi:10.1016/S0092-8674(00)81876-0.
- ↑ Longhese, MP; Bonetti, D; Guerini, I; Manfrini, N; Clerici, M (September 2009). "DNA double-strand breaks in meiosis: Checking their formation, processing and repair.". DNA Repair 8 (9): 1127–1138. doi:10.1016/j.dnarep.2009.04.005.
- ↑ "A DNA recombination "hotspot" in humans is missing in chimps". PLoS Biology 2 (6): e192. June 2004. PMC 423159. doi:10.1371/journal.pbio.0020192.
- ↑ Alberts B; et al. (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 303. ISBN 978-0-8153-4105-5.
- ↑ 22,0 22,1 Shrivastav, M; De Haro, LP; Nickoloff, JA (January 2008). "Regulation of DNA double-strand break repair pathway choice". Cell Research 18 (1): 134–147. PMID 18157161. doi:10.1038/cr.2007.111.
- ↑ 23,0 23,1 Mimitou, EP; Symington, LS (May 2009). "Nucleases and helicases take center stage in homologous recombination". Trends in Biochemical Science 34 (5): 264–272. PMID 19375328. doi:10.1016/j.tibs.2009.01.010.
- ↑ Huertas, P; Cortés-Ledesma, F; Sartori, AA; Aguilera, A; Jackson, SP (2 October 2008). "CDK targets Sae2 to control DNA-end resection and homologous recombination". Nature 455 (7213): 689–692. PMC 2635538. PMID 18716619. doi:10.1038/nature07215.
- ↑ 25,0 25,1 25,2 25,3 Sung, P; Klein, H (October 2006). "Mechanism of homologous recombination: mediators and helicases take on regulatory functions". Nature Reviews Molecular Cell Biology 7 (10): 739–750. PMID 16926856. doi:10.1038/nrm2008.
- ↑ Wold, MS (1997). "Replication protein A: heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism". Annual Review of Biochemistry 66: 61–92. PMID 9242902. doi:10.1146/annurev.biochem.66.1.61.
- ↑ McMahill, MS; Sham, CW; Bishop, DK (November 2007). "Synthesis-dependent strand annealing in meiosis". PLoS Biology 5 (11): e299. PMC 2062477. PMID 17988174. doi:10.1371/journal.pbio.0050299.
- ↑ Bärtsch, S; Kang, L; Symington, LS (February 2000). "RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates". Mol Cell Biol 20 (4): 1194–1205. PMC 85244. PMID 10648605. doi:10.1128/MCB.20.4.1194-1205.2000.
- ↑ Alberts B; et al. (2008). Molecular Biology of the Cell (5th ed.). Garland Science. pp. 312–313. ISBN 978-0-8153-4105-5.
- ↑ 30,0 30,1 30,2 Helleday, T; Lo, J; Van Gent, DC; Engelward, BP (1 July 2007). "DNA double-strand break repair: from mechanistic understanding to cancer treatment". DNA Repair (Amst.) 6 (7): 923–935. PMID 17363343. doi:10.1016/j.dnarep.2007.02.006.
- ↑ Haber lab. "Single-strand annealing". "Brandeis University". Arquivado dende o orixinal o 19 de xaneiro de 2015. Consultado o 3 July 2010.
- ↑ 32,0 32,1 32,2 Lyndaker, AM; Alani, E (March 2009). "A tale of tails: insights into the coordination of 3 end processing during homologous recombination". BioEssays 31 (3): 315–321. PMC 2958051. PMID 19260026. doi:10.1002/bies.200800195.
- ↑ Mimitou, EP; Symington, LS (September 2009). "DNA end resection: Many nucleases make light work". DNA Repair 8 (9): 983–995. PMC 2760233. PMID 19473888. doi:10.1016/j.dnarep.2009.04.017.
- ↑ Pâques, F; Haber, JE (June 1999). "Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae". Microbiology and molecular biology reviews : MMBR 63 (2): 349–404. PMC 98970. PMID 10357855.
- ↑ 35,0 35,1 McEachern, MJ; Haber, JE (2006). "Break-induced replication and recombinational telomere elongation in yeast". Annual Review of Biochemistry 75: 111–135. PMID 16756487. doi:10.1146/annurev.biochem.74.082803.133234.
- ↑ Morrish, TA; Greider, CW; Haber, James E. (January 2009). Haber, James E., ed. "Short telomeres initiate telomere recombination in primary and tumor cells". PLoS Genetics 5 (1): e1000357. PMC 2627939. PMID 19180191. doi:10.1371/journal.pgen.1000357.
- ↑ Muntoni, A; Reddel, RR (October 2005). "The first molecular details of ALT in human tumor cells". Human Molecular Genetics 14 (Review Issue 2): R191–R196. PMID 16244317. doi:10.1093/hmg/ddi266.
- ↑ Kowalczykowski, SC; Dixon, DA; Eggleston, AK; Lauder, SD; Rehrauer, WM (September 1994). "Biochemistry of homologous recombination in Escherichia coli". Microbiology and Molecular Biology Reviews 58 (3): 401–465. PMC 372975. PMID 7968921.
- ↑ Rocha, EPC; Cornet, E; Michel, B (August 2005). "Comparative and evolutionary analysis of the bacterial homologous recombination systems". PLoS Genetics 1 (2): e15. PMC 1193525. PMID 16132081. doi:10.1371/journal.pgen.0010015.
- ↑ 40,0 40,1 Amundsen, SK (15 December 2007). "Intersubunit signaling in RecBCD enzyme, a complex protein machine regulated by Chi hot spots". Genes Dev 21 (24): 3296–3307. PMC 2113030. PMID 18079176. doi:10.1101/gad.1605807.
- ↑ Singleton, MR; Dillingham, MS; Gaudier, M; Kowalczykowski, SC; Wigley, DB (11 November 2004). "Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks" (PDF). Nature 432 (7014): 187–193. PMID 15538360. doi:10.1038/nature02988. Arquivado dende o orixinal (PDF) o 25 de maio de 2004. Consultado o 26 de setembro de 2013.
- ↑ 42,0 42,1 Smith, GR (2012). "How RecBCD Enzyme and Chi Promote DNA Break Repair and Recombination: a Molecular Biologist's View". Microbiol Mol Biol Rev 76 (2): 217–28. PMID 22688812. doi:10.1128/MMBR.05026-11.
- ↑ 43,0 43,1 43,2 43,3 Dillingham, MS; Kowalczykowski, SC (December 2008). "RecBCD enzyme and the repair of double-stranded DNA breaks". Microbiology and Molecular Biology Reviews 72 (4): 642–671. PMC 2593567. PMID 19052323. doi:10.1128/MMBR.00020-08.
- ↑ Michel, B; Boubakri, H; Baharoglu, Z; LeMasson, M; M, R; Lestini, R (1 July 2007). "Recombination proteins and rescue of arrested replication forks". DNA Repair 6 (7): 967–980. PMID 17395553. doi:10.1016/j.dnarep.2007.02.016.
- ↑ Amundsen, SK; Taylor, AF; Reddy, M; Smith, G. R.; Smith, GR (December 2007). "Intersubunit signaling in RecBCD enzyme, a complex protein machine regulated by Chi hot spots". Genes Dev 21 (24): 3296–307. PMC 2113030. PMID 18079176. doi:10.1101/gad.1605807.
- ↑ Spies, M; Bianco, PR; Dillingham, MS; Handa, N; Baskin, RJ; Kowalczykowski, SC (5 September 2003). "A molecular throttle: the recombination hotspot Chi controls DNA translocation by the RecBCD helicase". Cell 114 (5): 647–654. PMID 13678587. doi:10.1016/S0092-8674(03)00681-0.
- ↑ Taylor, AF; Smith, GR (June 2003). "RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity". Nature 423 (6942): 889–93. PMID 12815437. doi:10.1038/nature01674.
- ↑ Spies, M; Amitani, I; Baskin, RJ; Kowalczykowski, Stephen C.; Kowalczykowski, SC (November 2007). "RecBCD enzyme switches lead motor subunits in response to Chi recognition". Cell 131 (4): 694–705. PMC 2151923. PMID 18022364. doi:10.1016/j.cell.2007.09.023.
- ↑ Savir Y & Tlusty T (2010). "RecA-mediated homology search as a nearly optimal signal detection system" (PDF). Molecular Cell 40 (3): 388–96. PMID 21070965. doi:10.1016/j.molcel.2010.10.020. Arquivado dende o orixinal (PDF) o 07 de outubro de 2012. Consultado o 26 de setembro de 2013.
- ↑ Rambo RP, Williams GJ, Tainer JA. (2010). "Achieving fidelity in homologous recombination despite extreme complexity: informed decisions by molecular profiling" (PDF). Molecular Cell 40 (3): 347–48. PMC 3003302. PMID 21070960. doi:10.1016/j.molcel.2010.10.032. Arquivado dende o orixinal (PDF) o 07 de outubro de 2012. Consultado o 26 de setembro de 2013.
- ↑ De Vlaminck I,van Loenhout MTJ, Zweifel L, den Blanken J, Hooning K, Hage S, Kerssemakers J, Dekker C (2012). "Mechanism of Homology Recognition in DNA Recombination from Dual-Molecule Experiments". Molecular Cell 46 (5): 616–624. doi:10.1016/j.molcel.2012.03.029.
- ↑ Alberts B; et al. (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 307. ISBN 978-0-8153-4105-5.
- ↑ Morimatsu, K; Kowalczykowski, SC (22 May 2003). "RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair". Molecular Cell 11 (5): 1337–1347. PMID 12769856. doi:10.1016/S1097-2765(03)00188-6.
- ↑ Hiom, K (July 2009). "DNA Repair: Common Approaches to Fixing Double-Strand Breaks". Current Biology 19 (13): R523–R525. PMID 19602417. doi:10.1016/j.cub.2009.06.009.
- ↑ Handa, N; Morimatsu, K; Lovett, ST; Kowalczykowski, SC (15 May 2009). "Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli". Genes & Development 23 (10): 1234–1245. PMC 2685532. PMID 19451222. doi:10.1101/gad.1780709.
- ↑ 56,0 56,1 West, SC (July 2003). "Molecular views of recombination proteins and their control". Nature Reviews Molecular Cell Biology 4 (6): 435–437. PMID 12778123. doi:10.1038/nrm1127.
- ↑ 57,0 57,1 57,2 57,3 Watson, JD; et al. (2003). Molecular Biology of the Gene (5th ed.). Pearson/Benjamin Cummings. pp. 259–291. ISBN 978-0-8053-4635-0.
- ↑ Gumbiner-Russo, LM; Rosenberg, SM; Sandler, Steve (28 November 2007). Sandler, Steve, ed. "Physical analyses of E. coli heteroduplex recombination products in vivo: on the prevalence of 5′ and 3′ patches". PLoS ONE 2 (11): e1242. PMC 2082072. PMID 18043749. doi:10.1371/journal.pone.0001242.
- ↑ Thomas, CM; Nielson, KM (September 2005). "Mechanisms of, and barriers to, horizontal gene transfer between bacteria" (PDF). Nature Reviews Microbiology 3 (9): 711–721. PMID 16138099. doi:10.1038/nrmicro1234. Arquivado dende o orixinal (PDF) o 01 de xuño de 2010. Consultado o 26 de setembro de 2013.
- ↑ Vulic, M; Dionisio, F; Taddei, F; Radman, M (2 September 1997). "Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria". Proceedings of the National Academy of Sciences USA 94 (18): 9763–9767. PMC 23264. PMID 9275198. doi:10.1073/pnas.94.18.9763.
- ↑ Majewski, J; Cohan, FM (January 1998). "The effect of mismatch repair and heteroduplex formation on sexual isolation in Bacillus". Genetics 48 (1): 13–18. PMC 1459767. PMID 9475717.
- ↑ Majewski, J; Zawadzki, P; Pickerill, P; Cohan, FM; Dowson, CG (February 2000). "Barriers to genetic exchange between bacterial species: Streptococcus pneumoniae transformation". Journal of Bacteriology 182 (4): 1016–1023. PMC 94378. PMID 10648528. doi:10.1128/JB.182.4.1016-1023.2000.
- ↑ Fleischmann Jr, WR (1996). "43". Medical Microbiology (4th ed.). University of Texas Medical Branch at Galveston. ISBN 0-9631172-1-1.
- ↑ Boni, MF; de Jong, MD; van Doorn, HR; Holmes, EC; Martin, Darren P. (3 May 2010). Martin, Darren P., ed. "Guidelines for identifying homologous recombination events in influenza a virus". PLoS ONE 5 (5): e10434. PMC 2862710. PMID 20454662. doi:10.1371/journal.pone.0010434.
- ↑ 65,0 65,1 Nagy, PD; Bujarski, JJ (January 1996). "Homologous RNA recombination in brome mosaic virus: AU-rich sequences decrease the accuracy of crossovers". Journal of Virology 70 (1): 415–426. PMC 189831. PMID 8523555.
- ↑ 66,0 66,1 Roossinck, MJ (September 1997). "Mechanisms of plant virus evolution". Annual Review of Phytopathology 35: 191–209. PMID 15012521. doi:10.1146/annurev.phyto.35.1.191.
- ↑ Arbuckle, Jesse (2011). "The molecular biology of human herpesvirus-6 latency and telomere integration". Microbes Infect. 13 (8-9): 731–41. PMC 3130849. PMID 21458587. doi:10.1016/j.micinf.2011.03.006.
- ↑ Lamb, NE; Yu, K; Shaffer, J; Feingold, E; Sherman, SL (January 2005). "Association between maternal age and meiotic recombination for trisomy 21". The American Journal of Human Genetics 76 (1): 91–99. PMC 1196437. PMID 15551222. doi:10.1086/427266.
- ↑ Cold Spring Harbor Laboratory (2007). "Human RecQ Helicases, Homologous Recombination And Genomic Instability". ScienceDaily. Consultado o 3 July 2010.
- ↑ Modesti, M; Kanaar, R (2001). "Homologous recombination: from model organisms to human disease". Genome Biology 2 (5): REVIEWS1014. PMC 138934. PMID 11387040. doi:10.1186/gb-2001-2-5-reviews1014.
- ↑ Luo, G; Santoro, IM; McDaniel, LD; Nishijima, I; Mills, M; Youssoufian, H; Vogel, H; Schultz, RA; Bradley, A (December 2000). "Cancer predisposition caused by elevated mitotic recombination in Bloom mice". Nature Genetics 26 (4): 424–429. PMID 11101838. doi:10.1038/82548.
- ↑ 72,0 72,1 Alberts, B; et al. (2007). Molecular Biology of the Cell (5th ed.). Garland Science. ISBN 978-0-8153-4110-9.
- ↑ 73,0 73,1 73,2 Powell, SN; Kachnic, LA (September 2003). "Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation". Oncogene 22 (37): 5784–5791. PMID 12947386. doi:10.1038/sj.onc.1206678.
- ↑ 74,0 74,1 74,2 74,3 74,4 74,5 Lin, Z; Kong, H; Nei, M; Ma, H (5 July 2006). "Origins and evolution of the recA/Rad51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer". Proceedings of the National Academy of Sciences USA 103 (27): 10328–10333. PMC 1502457. PMID 16798872. doi:10.1073/pnas.0604232103.
- ↑ Jain, SK; Cox, MM; Inman, RB (12 August 1994). "On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. III. Unidirectional branch migration and extensive hybrid DNA formation". Journal of Biological Chemistry 269 (32): 20653–20661. PMID 8051165.
- ↑ Ramesh, MA; Malik, SB; Logsdon Jr, JM (26 January 2005). "A phylogenomic inventory of meiotic genes; evidence for sex in Giardia and an early eukaryotic origin of meiosis". Current Biology 15 (2): 185–191. PMID 15668177. doi:10.1016/j.cub.2005.01.003.
- ↑ 77,0 77,1 Malik, SB; Ramesh, MA; Hulstrand, AM; Logsdon Jr, JM (December 2007). "Protist homologs of the meiotic Spo11 gene and topoisomerase VI reveal an evolutionary history of gene duplication and lineage-specific loss". Molecular Biology and Evolution 24 (12): 2827–2841. PMID 17921483. doi:10.1093/molbev/msm217.
- ↑ Lodish H; et al. (2000). "8.5:Gene Replacement and Transgenic Animals: DNA Is Transferred into Eukaryotic Cells in Various Ways". Molecular Cell Biology (4th ed.). W. H. Freeman and Company. ISBN 0-7167-3136-3.
- ↑ "The Nobel Prize in Physiology or Medicine 2007". The Nobel Foundation. Consultado o December 15, 2008.
- ↑ Drummond, DA; Silberg, JJ; Meyer, MM; Wilke, CO; Arnold, FH (12 April 2005). "On the conservative nature of intragenic recombination". Proceedings of the National Academy of Sciences USA 102 (15): 5380–5385. PMC 556249. PMID 15809422. doi:10.1073/pnas.0500729102.
- ↑ Bloom, JD; Silberg, JJ; Wilke, CO; Drummond, DA; Adami, C; Arnold, FH (15 January 2005). "Thermodynamic prediction of protein neutrality". Proceedings of the National Academy of Sciences USA 102 (3): 606–611. PMC 545518. PMID 15644440. doi:10.1073/pnas.0406744102.
- ↑ 82,0 82,1 Carbone, MN; Arnold, FH (August 2007). "Engineering by homologous recombination: exploring sequence and function within a conserved fold". Current Opinion in Structural Biology 17 (4): 454–459. PMID 17884462. doi:10.1016/j.sbi.2007.08.005.
- ↑ Otey, CR; Landwehr, M; Endelman, JB; Hiraga, K; Bloom, JD; Arnold, FH (May 2006). "Structure-guided recombination creates an artificial family of cytochromes P450". PLoS Biology 4 (5): e112. PMC 1431580. PMID 16594730. doi:10.1371/journal.pbio.0040112.
- ↑ Socolich, M; Lockless, SW; Russ, WP; Lee, H; Gardner, KH; Ranganathan, R (22 September 2005). "Evolutionary information for specifying a protein fold". Nature 437 (7058): 512–518. PMID 16177782. doi:10.1038/nature03991.
- ↑ Thulasiram, HV; Erickson, HK; Poulter, CD (6 April 2007). "Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis". Science 316 (5821): 73–76. PMID 17412950. doi:10.1126/science.1137786.
- ↑ Landwehr, M; Carbone, M; Otey, CR; Li, Y; Arnold, FH (March 2007). "Diversification of catalytic function in a synthetic family of chimeric cytochrome P450s". Chemistry and Biology 14 (3): 269–278. PMC 1991292. PMID 17379142. doi:10.1016/j.chembiol.2007.01.009.
- ↑ Nelson, DL; Cox, MM (2005). Principles of Biochemistry (4th ed.). Freeman. pp. 896. ISBN 978-0-7167-4339-2.
- ↑ 88,0 88,1 88,2 Iglehart, JD; Silver, DP (9 July 2009). "Synthetic lethality — a new direction in cancer-drug development". New England Journal of Medicine 361 (2): 189–191. PMID 19553640. doi:10.1056/NEJMe0903044.
- ↑ Fong, FC; Boss, DS; Yap, TA; Tutt, A; Wu, P; Mergui-Roelvink, M; Mortimer, P; Swaisland, H; Lau, A (9 July 2009). "Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers". New England Journal of Medicine 361 (2): 123–134. PMID 19553641. doi:10.1056/NEJMoa0900212.
- ↑ Edwards, SL; Brough, R; Lord, CJ; Natrajan, R; Vatcheva, R; Levine, DA; Boyd, J; Reis-Filho, JS; Ashworth, A (28 February 2008). "Resistance to therapy caused by intragenic deletion in BRCA2". Nature 451 (7182): 1111–1115. PMID 18264088. doi:10.1038/nature06548.
Véxase tamén
editarLigazóns externas
editar| Commons ten máis contidos multimedia sobre: Recombinación homóloga |
- Animacións – Recombinación homóloga: Animacións de varios modelos de recombinación homóloga.
- Recombinación homóloga: Tempy & Trun: Animación da vía recBCD bacteriana da recombinación homóloga