Estrutura primaria dos ácidos nucleicos
- Para as técnicas para a determinación da secuencia do ADN ver secuenciación do ADN.
A estrutura primaria dos ácidos nucleico é a sucesión de nucleótidos (unidades que forman os ácidos nucleicos) representados polas letras A, T, G, C, U, que indica a orde en que estes están situados na molécula. Unha secuencia de ácido nucleico ou de nucleótidos é esa mesma sucesión nun ADN (letras A, T, G, C) ou nun ARN (letras A, U, G, C). Por convención, as secuencias son xeralmente presentadas empezando polo extremo 5' e acabando polo extremo 3'. Como os ácidos nucleicos son xeralmente polímeros lineares (é dicir, non ramificados), especificar a súa secuencia equivale a definir a estrutura covalente da molécula enteira. Nesta estrutura primaria está codificada a información xenética dos seres vivos, polo que ás veces tamén se usa para ela a expresión secuencia xenética. As secuencias poden lerse a partir do material biolóxico utilizando diversos métodos de secuenciación do ADN.
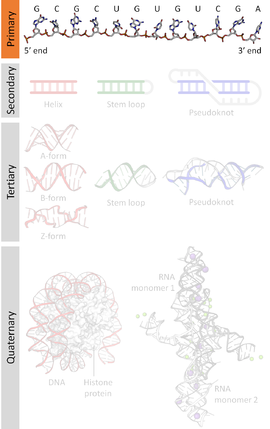

Ademais da estrutura primaria (non se debe dicir "secuencia primaria"), os ácidos nucleicos teñen estrutura secundaria, terciaria e superiores.
Nucleótidos editar

Os ácidos nucleicos están formados por unha sucesión de moléculas chamadas nucleótidos unidas entre si por un enlace covalente chamado fosfodiéster. Os nucleótidos están formados por un fosfato, un azucre e unha base nutroxenada. O fosfato nunca varía. O azucre serve para distinguir o ADN (leva o azucre desoxirribosa) do ARN (leva ribosa). A base nitroxenada (ou nucleobase) é o que distingue nun mesmo ácido nucleico un nucleótido doutro, polo que a secuencia dun ácido nucleico é tamén a súa secuencia de nucleótidos ou a súa secuencia de bases. Segundo as bases nitroxenadas que leven hai cinco tipos de nucleótidos habituais nos ácidos nucleicos, que son: adenina (representado por A), guanina (G), citosina (C), timina (T) e uracilo (U). No ADN aparecen A, T, G e C, e no ARN A, U, G e C. Nos ácidos nucleicos bicatenarios, como o ADN, as bases emparéllanse coas da cadea de enfronte formando sempre os pares A-T e G-C mediante o establecemento de pontes de hidróxeno (outros emparellamentos non terían os grupos químicos adecuados para formar esas pontes de hidróxeno). Isto quere dicir que unha cadea é complementaria en bases da outra.
Xeralmente unha secuencia de ADN escríbese coas bases seguidas (sen espazos entre elas) de esquerda a dereita desde o extremo 5' ao 3', por exemplo 5'-AAAGTCTGAC-3'. Durante a tradución de proteínas, os ARNm son traducidos empezando polo extremo 5'. Por exemplo, a secuencia complementaria de 5'-TTAC-3' é 5'-GTAA-3' (obsérvese que as dúas están escritas en sentido 5'-3', pero nun ADN bicatenario estarán colocadas de forma antiparalela, polo que o primeiro T da primeira secuencia terá en fronte o o último A da segunda secuencia, e así as demais).
Notación editar
Na representación das secuencias de nucleótidos A, T, C, G e U representan a adenina, timina, citosina, guanina e uracilo, pero hai tamén letras para representar a ambigüidade nunha determinada posición, que podería estar ocupada por un nucleótido entre varios posibles. As regras da IUPAC para estes casos son:[1]
- R = G A (unha purina)
- Y = T C (unha pirimidina)
- K = G T (ceto)
- M = A C (amino)
- S = G C (enlazamento forte, con tres pontes de hidróxeno)
- W = A T (enlazamento feble, con dúas pontes de hidróxeno)
- B = G T C (todos excepto A)
- D = G A T (todos menos C)
- H = A C T (todos agás G)
- V = G C A (todos excepto T)
- N = A G C T (calquera)
Estes símbolos son tamén válidos para o ARN, pero nese caso U (uracilo) substituiría a T (timina).[1]
Ademais das bases habituais A, C, G, T e U, os ácidos nucleicos poden tamén conter bases modificadas despois de que se formou a cadea do ácido nucleico. No ADN, a base modificada máis común é a 5-metilcitidina (m5C). No ARN, pode haber moitas bases modificadas, como a pseudouridina (Ψ), dihìdrouridina (D), inosina (I), ribotimidina (rT) e 7-metilguanosina (m7G).[2][3] A hipoxantina e a xantina son dúas das bases que se poden orixinar pola presenza de mutáxenos.[4] De xeito similar, a desaminación da citosina orixina uracilo.
Importancia biolóxica editar

- Véxase tamén: Código xenético e Dogma central da bioloxía molecular.
Nos sistemas biolóxicos, os ácidos nucleicos conteñen a información que utiliza unha célula viva para construír as súas proteínas específicas. A secuencia de nucleobases dunha cadea de ácido nucleico é traducida pola maquinaria celular nunha secuencia de aminoácidos formando unha cadea proteica. Cada grupo de tres bases, chamado codón, corresponde a un determinadao aminoácido, e existe un código xenético específico no que se asigna a cada posible combinación de tres bases un determinado aminoácido.
O dogma central da bioloxía molecular indica a dirección en que flúe a información xenética desde o ADN ata as proteínas. Primeiro, o ADN transcríbese a moléculas de ARNm, que se dirixen ao ribosoma, onde ese ARNm será utilizado como molde para a construción da proteína.
As mutacións causan cambios nos nucleótidos e, por tanto, nos codóns, polo que estas alteracións da secuencia do ADN dan lugar a que se formen proteínas con secuencias de aminoácidos distintas da nornal, que poden ser non funcionais. Algunhas mutacións poden facer que non sexa posible fabricar a proteína.
A secuenciación de xenomas enteiros ten grande importancia para establecer as relacións filoxenéticas dos seres vivos. Secuenciáronse os xenomas de moitos microorganismos e duns cantos eucariotas. Un dos secuenciados foi o xenoma humano. É tamén importante o estudo de polimorfismos dun só nucleótido ou de indeis sinatura conservados e outros marcadores xenéticos.
Determinación da secuencia editar

- Artigo principal: secuenciación do ADN.
A secuenciación do ADN é o proceso de determinar a secuencia de nucleótidos dun fragmento de ADN dado. Como a secuencia do ADN dun ser vivo codifica a información necesaria para ese ser vivo, determinar a súa secuencia é útil na investigación fundamental de como funcionan os organismos, ou determinados individuos, e en practicamente todas as investigacións biolóxicas. Por exemplo, en medicina pode utilizarse para identificar, diagnosticar e potencialmente desenvolver tratamentos para as enfermidades xenéticas. De igual xeito, a investigación dos patóxenos pode levar a idear tratamentos para as enfermidades contaxiosas. A biotecnoloxía é unha disciplina florecente e con gran potencial, que utiliza moito a secuenciación do ADN.
O ARN non se secuencia directamente, senón que é copiado primeiro a ADN utilizando o encima transcriptase inversa, e é ese ADN o que se secuencia.
Os métodos de secuenciación actuais dependen da capacidade discriminatoria das ADN polimerases, e, por tanto, só poden distinguir catro bases. Unha inosina (creada a partir de adenosina durante a edición do ARN (RNA editing) lese como G, e a 5-metilcitosina (creada a partir da citosina por metilación do ADN) lese como C. Coa tecnoloxía actual, é difícil secuenciar pequenas cantidades de ADN, xa que o sinal é demasiado feble para medilo, pero iso pódese solucionar facendo primeiro unha amplificación do ADN coa técnica da reacción en cadea da polimerase (PCR).
Representación dixital editar

Unha vez que se obtivo unha secuencia de ácido nucleico dun organismo esta almacénase en formato dixital. As secuencias xenéticas dixitais poden ser almacenadas en bases de datos de secuencias, ser analizadas (ver Análise de secuencias máis abaixo), ser alteradas dixitalmente e utilizadas como moldes para crear novas moléculas de ADN reais utilizando a síntese de xenes artificial.
Análise de secuencias editar
- Artigo principal: Análise de secuencias.
As secuencias xenéticas dixitais poden ser analizadas utilizando as ferramentas bioinformáticas para intentar determinar a súa función.
Probas xenéticas editar
O ADN do xenoma dos organismos pode ser analizado para diagnosticar a susceptibilidade a ter enfermidades conxénitas, e pode tamén utilizarse para determinar a paternidade dun neno (pai xenético) ou os devanceiros dunha persoa. Normalmente, cada persoa leva dúas copias de cada xene, unha herdada da nai e outra do pai, e estas copias poden ser iguais ou pertencer a variedades distintas (alelos) dese xene. O xenoma humano crese que contén arredor de 20.000 - 25.000 xenes. Ademais as probas xenéticas en senso amplo inclúen probas bioquímicas para a posible presenza de enfermidades xenéticas, ou formas mutantes de xenes asociados co incremento do risco de desenvolver trastornos xenéticos.
As probas xenéticas identifican cambios nos cromosomas, xenes, ou proteínas.[5] Xeralmente, as probas utilízanse para detectar cambios que están asociados con trastornos herdados e poden confirmar ou excluír unha condición xenética sospeitada e determinar a probabilidade de desenvolvela ou transmitila á descendencia.[6][7]
Aliñamento de secuencias editar
- Artigo principal: Aliñamento de secuencias.
En bioinformática, un aliñamento de secuencias é un modo de dispoñer as secuencias de ADN, ARN, ou proteína para identificar rexións semellantes que poden selo debido a relacións funcionais, estruturais, ou evolutivas entre as secuencias.[8] Se dúas secuencias dun aliñamento comparten un antepasado común, as faltas de correspondencia poden interpretarse como mutacións puntuais e as lagoas (gaps) como mutacións por inserción ou deleción (indeis) introducidos nunha ou en ambas as liñaxes no período de tempo que pasou desde que diverxiron unha da outra. Nos aliñamentos de secuencias de proteínas, o grao de semellanza entre aminoácidos que ocupan unha determinada posición na secuencia pode ser interpretada como unha medida grosso modo do conservada que está unha determinada rexión ou secuencia motivo entre distintas liñaxes. A ausencia de substitucións, ou a presenza unicamente de substitucións moi conservadoras (é dicir, a substitución de aminoácidos cuxas cadeas laterais teñen propiedades bioquímicas similares) nunha determinada rexión da secuencia, suxiren[9] que esta rexión ten unha importancia estrutural ou funcional. Aínda que as bases nitroxenadas do ADN e ARN son máis similares entre si que cos aminoácidos, a conservación dos pares de bases pode indicar un papel funcional ou estrutural similar.
A filoxenética computacional fai un amplo uso dos aliñamentos de secuencia na construción e interpretación de árbores filoxenéticas, que se utilizan para clasificar as relacións evolutivas entre xenes homólogos representados nos xenomas de especies diverxentes. O grao no cal as secuencias dun conxunto estudado difiren está cualitativamente relacionado coa distancia evolutiva entre as secuencias. En xeral, unha alta identidade de secuencia indica que as secuencias en cuestión teñen un antepasado común máis recente comparativamente recente, mentres que unha baixa identidade indica que a diverxencia é máis antiga. Esta aproximación, que reflicte a hipótese do "reloxo molecular" de que se pode usar un taxa case constante de cambio evolutivo para extrapolar o tempo que pasou desde que diverxiron dous xenes (é dicir, o tempo de coalescencia), asume que os efectos da mutación e a selección natural son constantes entre as liñaxes. Por tanto, non considera posibles diferenzas entre organismos ou especies nas taxas de reparación do ADN ou a posible conservación funcional de rexións específicas dunha secuencia. (No caso das secuencias de nucleótidos, a hipótese do reloxo molecular na súa forma máis básica tampouco ten en conta as diferenzas nas taxas de aceptación entre mutacións silenciosas que non alteran o significado dun determinado codón e outras mutacións que dan lugar á incorporación dun aminoácido diferente nas proteína)s. Outros métodos estatísticos máis exactos permiten que varíe a taxa de evolución en cada póla da árbore filoxenética, producindo así mellores estimacións dos tempos de coalescencia para os xenes.
Secuencia motivo editar
- Artigo principal: Secuencia motivo.
Frecuentemente, a estrutura primaria codifica motivos que teñen importancia funcional. Algúns exemplos de secuencias motivo son: a caixa C/D[10] e a caixa H/ACA[11] dos snoRNAs, sitio de unión Sm que se encontra nos ARNs do espliceosoma como o U1, U2, U4, U5, U6, U12 e U3, a secuencia Shine-Dalgarno,[12] a secuencia consenso Kozak[13] e a do terminador da ARN polimerase III.[14]
Notas editar
- ↑ 1,0 1,1 Nomenclature for Incompletely Specified Bases in Nucleic Acid Sequences, NC-IUB, 1984.
- ↑ "BIOL2060: Translation". Arquivado dende o orixinal o 25 de xullo de 2017. Consultado o 08 de xullo de 2013.
- ↑ Research
- ↑ T Nguyen, D Brunson, C L Crespi, B W Penman, J S Wishnok, and S R Tannenbaum, DNA damage and mutation in human cells exposed to nitric oxide in vitro, Proc Natl Acad Sci U S A. 1992 April 1; 89(7): 3030–3034
- ↑ "What is genetic testing? - Genetics Home Reference". Arquivado dende o orixinal o 29 de maio de 2006. Consultado o 08 de xullo de 2013.
- ↑ Genetic Testing: MedlinePlus
- ↑ "Definitions of Genetic Testing". Definitions of Genetic Testing (Jorge Sequeiros and Bárbara Guimarães). EuroGentest Network of Excellence Project. 2008-09-11. Arquivado dende o orixinal o 23 de maio de 2018. Consultado o 2008-08-10.
- ↑ Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.). Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY. ISBN 0-87969-608-7.
- ↑ Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001 May;11(5):863-74.
- ↑ Samarsky, DA; Fournier MJ, Singer RH, Bertrand E (1998). "The snoRNA box C/D motif directs nucleolar targeting and also couples snoRNA synthesis and localization". EMBO 17 (13): 3747–3757. PMC 1170710. PMID 9649444. doi:10.1093/emboj/17.13.3747.
- ↑ Ganot, Philippe; Caizergues-Ferrer, Michèle; Kiss, Tamás (1 April 1997). "The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation". Genes & Development 11 (7): 941–956. doi:10.1101/gad.11.7.941. PMID 9106664.
- ↑ Shine J, Dalgarno L (1975). "Determinant of cistron specificity in bacterial ribosomes". Nature 254 (5495): 34–8. PMID 803646. doi:10.1038/254034a0.
- ↑ Kozak M (1987). "An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs". Nucleic Acids Res. 15 (20): 8125–8148. PMC 306349. PMID 3313277. doi:10.1093/nar/15.20.8125.
- ↑ Bogenhagen DF, Brown DD (1981). "Nucleotide sequences in Xenopus 5S DNA required for transcription termination.". Cell 24 (1): 261–70. PMID 6263489. doi:10.1016/0092-8674(81)90522-5.
Véxase tamén editar
Outros artigos editar
- Polimorfismo dun só nucleótido (SNP)
- Análises moleculares de ADN
- Direccionalidade (bioloxía molecular)
- Sentido (bioloxía molecular)