Microscopio de fluorescencia
Un microscopio de fluorescencia é un microscopio óptico que usa a fluorescencia en lugar de (ou ademais de) a dispersión, a reflexión e a atenuación ou absorción, para estudar as propiedades de substancias orgánicas ou inorgánicas.[1][2] Pode denominarse microscopio de fluorescencia a calquera microscopio que use a fluorescencia para xerar unha imaxe, tanto se é un aparello simple, como un microscopio de epifluorescencia, coma se ten un deseño máis complicado, como un microscopio confocal, que usa o seccionamento óptico para conseguir unha mellor resolución da imaxe fluorescente.[3] A microscopia de fluorescencia utilízase moito para obter imaxes de mostras biolóxicas.

Principio
editarO espécime ilumínase con luz dunha lonxitude (ou lonxitudes) de onda específicas, que son absorbidas polos fluoróforos, causando que emitan luz de lonxitudes de onda máis longas (é dicir, dunha cor diferente á da luz absorbida). A luz usada para a iluminación é separada da fluorescencia moito máis feble emitida utilizando un filtro de emisión espectral. Os compoñentes típicos dun microscopio de fluorescencia son unha fonte de luz (a lámpada de arco de xenon ou a lámpada de vapor de mercurio son comúns; as formas máis avanzadas son LEDs de alta potencia e láseres), o filtro de excitación, o espello dicroico (ou divisor do raio dicroico), e o filtro de emisión (ver figura máis abaixo). Os filtros e o divisor do raio dicroico elíxense para que se correspondan coa excitación espectral e as características de emisión do fluoróforo usado para etiquetar o espécime.[1] Desta maneira, visualízase a distribución dun só fluoróforo (cor) á vez. Deben compoñerse as imaxes de moitas cores de varios tipos de fluoróforos combinando varias imaxes dunha soa cor.[1]
A maioría dos microscopios de fluorescencia en uso son microscopios de epifluorescencia, onde a excitación do fluoróforo e a detección da fluorescencia fanse na mesma vía de luz (é dicir, a través do obxectivo). Estes microscopios son moi utilizados en bioloxía e neles baséanse os deseños de microscopios máis avanados, como o microscopio confocal e o microscopio de fluorescencia de reflexión interna total (TIRF).
Microscopia de epifluorescencia
editar
A maioría dos microscopios de fluorescencia, especialmente os usados nas ciencias da vida, son os de deseño de epifluorescencia mostrados no diagrama. A lonxitude de onda da luz de excitación ilumina o espécime a través da lente obxectivo. A fluorescencia emitida polo espécime enfócase no detector polo mesmo obxectivo que se usa para a excitación, o cal para unha maior resolución necesitará unha lente obxectivo cunha maior apertura numérica. Como a maioría da luz de excitación se transmite a través do espécime, só a luz excitatoria chega ao obxectivo xunto coa luz emitida e a método epifluorescente dá, por tanto, unha alta razón sinal-ruído. O divisor do raio dicroico actúa como un filtro específico de lonxitude de onda, transmitindo luz de fluorescencia a través do ocular ou detector, pero reflectindo calquera outra luz de excitación restante de volta á fonte.
Fontes de luz
editarA microscopia de fluorescencia require unha iluminación intensa, case monocromática que algunhas fontes de luz moi utilizadas, como as lámpadas halóxenas, non poden proporcionar.[4] Utilízanse catro tipos principais de fontes de luz, incluíndo as lámpadas de arco de xenon ou lámpadas de vapor de mercurio cun filtro de excitación, láseres, fontes supercontinuas, e os LEDs de alta potencia. Os láseres son as fontes máis utilizadas nas técnicas de microscopia de fluorescencia complexa como a microscopia confocal e a microscopia de fluorescencia de reflexión interna total, mentres que as lámpadas de xenon, as de mercurio e os LEDs cun filtro de excitación dicroico utilízanse comunmente para os microscopios de epifluorescencia de campo ancho. Situando dous conxuntos de microlentes na vía de iluminación dun microscopio de epifluorescencia de campo ancho,[5] pode conseguirse unha iluminación moi uniforme cun coeficiente de variación do 1-2%.
Preparación da mostra
editarMostra transparencias superpostas de catro canles fluorescentes (a) Verde: [fluorescencia DiOC6(3)] - tingue membranas celulares que indican os corpos celulares centrais
(b) Ciano: [fluorescencia PLL-A546] - contratinguidura xenérica para visualizar superficies de células eucariotas
(c) Azul: [fluorescencia Hoechst] - tingue o ADN, identifica os núcleos
(d) Vermello: [antifluorescencia de clorofila] - resolve cloroplastos[6]
A animación empeza superpoñendo toda as canles fluorescentes dispoñibles e despois clarifica a visualización cambiando canles encendidas e apagadas.

Para que unha mostra sexa axeitada para utilizarse na microscopia de fluorescencia debe ser fluorescente. Hai varios métodos para crear unha mostra fluorescente; as principais técnicas son etiquetar con tinguiduras fluorescentes ou, no caso de mostras biolóxicas, a expresión dunha proteína fluorescente. Alternativamente, pode utilizarse a fluorescencia intrínseca dunha mostra (é dicir, a autofluorescencia).[1] A microscopia de fluorescencia nas ciencias da vida é unha poderosa ferramenta que permite a tinguidura específica e sensible dun espécime para detectar a distribución das proteínas ou outras moléculas de interese. Como resultado, hai un conxunto diverso de técnicas para a tinguidura fluorescente de mostras biolóxicas.
Tinguidura fluorescente biolóxica
editarMoitas tinguiduras fluorescentes foron deseñadas para un conxunto de moléculas biolóxicas. Algunhas destas son pequenas moléculas que son intrinsecamente fluorescentes e únense a moléculas biolóxicas de interese. Os principais exemplos destas tinguiduras son as tinguiduras para ácidos nucleicos como a DAPI e a Hoechst (excitadas pola luz UV) e as DRAQ5 e DRAQ7 (excitadas optimamente pola luz vermella), que todas elas se unen ao suco menor do ADN, etiquetando así os núcleos da célula. Outros son fármacos, toxinas, ou péptidos que se unen a estruturas celulares específicas e foron modificadas cun reporteiro fluorescente. Un exemplo importante desta clase de tinguidura fluorescente é a faloidina, que se usa para tinguir fibras de actina en células de mamíferos. Un novo péptido, coñecido como péptido que se hibrida ao coláxeno, pode tamén conxugarse con fluoróforos e usarse para tinguir fibras de coláxeno desnaturalizadas. A tinguidura das paredes celulares das plantas realízase utilizando marcaxes e tinguiduras que se unen á celulosa ou pectina. Continúa a busca de sondas fluorescentes cunha alta especificidade que tamén permitan obter imaxes de células vexetais vivas.[7]
Hai moitas moléculas fluorescencias chamadas fluoróforos ou fluorocromos como a fluoresceína, Alexa Fluors ou DyLight 488, que poden ligarse quimicamente a unha molécula diferente que se une á diana de interese dentro da mostra.
Inmunofluorescencia
editarA inmunofluorescencia é unha técnica que utiliza a unión altamente específica dun anticorpo ao seu antíxeno para etiquetar proteínas específicas ou outras moléculas dentro da célula. Unha mostra trátase cun anticorpo primario específico para a molécula de interese. Pode conxugarse un fluoróforo directamente ao anticorpo primario. Alternativamente, pode utilizarse un anticorpo secundario, conxugado a un fluoróforo, que se une especificamente ao primeiro anticorpo. Por exemplo, un anticorpo primario xerado nun rato que recoñece a tubulina combinado cun anticorpo secundario derivado cun fluoróforo podería usarse para etiquetar os microtúbulos da célula.
Proteínas fluorescentes
editarO coñecemento moderno da xenética e as técnicas dispoñibles para modificar o ADN permite aos científicos modificar xeneticamente proteínas para que tamén leven unha proteína fluorescente reporteira. En mostras biolóxicas isto permite facer directamente que unha proteína de interese sexa fluorescente. A localización proteica pode despois rastrearse directamente, incluso en células vivas.
Limitacións
editarOs fluoróforos perden a súa capacidade de fluorescencia a medida que son iluminados por un proceso chamado fotobranqueamento. O fotobranqueamento ocorre a medida que as moléculas fluorescentes acumulan danos químicos polos electróns excitados durante a fluorescencia. O fotobranqueamento pode limitar moito o tempo durante o cal unha mostra pode observarse con microscopia de fluorescencia. Existen varias técnicas que reducen o fotobranqueamento, como o uso de fluoróforos máis robustos, minimizar a iluminación ou usar compostos químicos fotoprotectores.
A microscopia de fluorescencia con proteínas reporteiras fluorescentes permitiu a análise de células vivas; porén, as células son susceptibles de fototoxicidade, especialmente con lonxitudes de onda curtas. Ademais, as moléculas fluorescentes teñen unha tendencia a xerar especies químicas reactivas cando están baixo iluminación que potencia o efecto fototóxico.
A diferenza das técnicas de microscopia de luz transmitida e reflectida, a microscopia de fluorescencia soamente permite a observación de estruturas específicas que foran etiquetadas para a fluorescencia. Por exemplo, observar unha mostra de tecido preparada cunha tinguidura fluorescente para o ADN por microscopia de fluorescencia só revela a organización do ADN do interior das células e non revela nada máis sobre a morfoloxía das células.
As técnicas computacionais que se propoñen estimar o sinal fluorescente de imaxes non fluorescentes (como o campo brillante) poden reducir estes problemas.[8] En xeral, estas estratexias implican adestrar unha rede neural convolucional profunda en células tinguidas e despois estimar a fluorescencia en mostras non tinguidas. Así, desacoplando as células baixo investigación das células usadas para adestrar a rede, as imaxes poden obterse máis rápido e cunha reducida fototoxicidade.
Técnicas de subdifracción
editarA natureza ondulatoria da luz limita o tamaño do punto sobre o cal se pode enfocar a luz debido ao límite de difracción. Esta limitación foi descrita no século XIX por Ernst Abbe e "limita a resolución dun microscopio óptico a aproximadamente a metade da lonxitude de onda da luz utilizada." A microscopia de fluorescencia é básica para moitas técnicas que teñen como obxectivo superar este límite con configuracións ópticas especializadas.
Inventáronse varias melloras en técnicas de microscopia no século XX que produciron un incremento na resolución e contraste ata certo punto. Porén, non puideron superar o límite de difracción. En 1978 desenvolvéronse as primeiras ideas teóricas para romper esta barreira usando un microscopio 4Pi, como un a microscopio de fluorescencia de varrido láser confocal no que a luz se enfoca idealmente desde todos os lados nun foco común que se utiliza para facer un varrido do obxecto por excitación "punto a punto" combinada cunha detección "punto a punto".[9] Porén, a primeira demostración experimental do microscopio 4pi tivo lugar en 1994.[10] A microscopia 4Pi maximiza a cantidade de direccións de enfoque dispoñibles usando dúas lentes obxectivo opostas ou microscopia de excitación de dous fotóns usando luz desprazada ao vermello e excitación multi-fotón.
A microscopia correlativa integrada combina un microscopio de fluorescencia cun microscopio electrónico. Isto permite ver a ultraestrutura e ter información contextual cun microscopio electrónico mentres se usan datos do microscopio de fluorescencia como ferramenta de etiquetado.[11]
A primeira técnica para conseguir realmente unha resolución subdifracción foi a microscopia STED, proposta en 1994. Este método e todas as técnicas que seguiron o concepto RESOLFT dependen dunha forte interacción non linear entre a luz e as moléculas que están emitindo fluorescencia. As moléculas son levadas entre estados moleculares distinguibles en cada localización específica, así que finalmente a luz pode emitirse a só unha pequena fracción do espazo, producindo un incremento da resolución.
Tamén na década de 1990, desenvolveuse outro método de microscopia de super-resolución baseado na microscopia de campo ancho. Conseguiuse unha mellora substancial do tamaño de resolución de nanoestruturas celulares tinguidas cun marcador fluorescente desenvolvendo a microscopia de localización SPDM e a iluminación láser estruturada (iluminación modulada especialmente ou SMI polas súas siglas en inglés).[12] Combinando o principio do SPDM coa SMI conseguiuse o desenvolvemento do microscopio Vertico SMI.[13][14] A detección de moléculas únicas de marcadores fluorescentes palpebreantes normais, como a proteína fluorescente verde (GFP) pode conseguirse usando un ulterior desenvolvemento do SPDM denominado tecnoloxía SPDMphymod, que fai posible detectar e contar dous tipos de moléculas fluorescentes a nivel molecular (esta tecnoloxía denomínase microscopia de localización de dúas cores ou 2CLM).[15]
Alternativamente, o advimento da microscopia de localización fotoactivada podería conseguir resultados similares baseándose no palpebreo ou cambio de moléculas únicas, no que a fracción de moléculas fluorescentes é moi pequena en cada momento. Esta resposta estocástica das moléculas á luz aplicada corresponde tamén a unha interacción altamente non linear, que leva a unha resolución subdifracción.
Galería de micrografías de fluorescencia
editar-

-
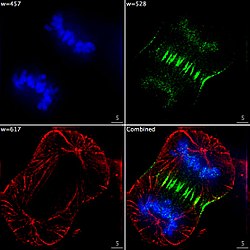 Imaxe epifluorescente de tres compoñentes dunha célula cancerosa humana en división. O ADN está marcado de azul, unha proteína chamada INCENP é verde, e os microtúbulos son vermellos. Cada fluoróforo foi captado separadamente usando unha combinación diferente de filtros de emisión e excitación, e as imaxes foron tomadas secuencialmente usando unha cámara CCD dixital, superpoñéndoas despois para obter unha imaxe completa.
Imaxe epifluorescente de tres compoñentes dunha célula cancerosa humana en división. O ADN está marcado de azul, unha proteína chamada INCENP é verde, e os microtúbulos son vermellos. Cada fluoróforo foi captado separadamente usando unha combinación diferente de filtros de emisión e excitación, e as imaxes foron tomadas secuencialmente usando unha cámara CCD dixital, superpoñéndoas despois para obter unha imaxe completa. -

-
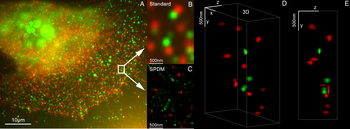 Microscopia de super-resolución de cor dual 3D con Her2 e Her3 en células mamarias, tinguiduras estándar: Alexa 488, Alexa 568. Microscopia LIMON.
Microscopia de super-resolución de cor dual 3D con Her2 e Her3 en células mamarias, tinguiduras estándar: Alexa 488, Alexa 568. Microscopia LIMON. -
 Núcleos de linfocitos humanos tinguidos con DAPI con sondas hibridadas nos centrómeros dos cromosomas 13 (verde) e 21 (vermello) (hibridación in situ fluorescente (FISH)).
Núcleos de linfocitos humanos tinguidos con DAPI con sondas hibridadas nos centrómeros dos cromosomas 13 (verde) e 21 (vermello) (hibridación in situ fluorescente (FISH)). -

-
 Microscopia de super.resolución: Detección dunha molécula de YFP única nunha célula de cancro humana. As medidas de distancia típicas están no rango dos 15 nm (medido cun microscopio Vertico-SMI/SPDMphymod).
Microscopia de super.resolución: Detección dunha molécula de YFP única nunha célula de cancro humana. As medidas de distancia típicas están no rango dos 15 nm (medido cun microscopio Vertico-SMI/SPDMphymod). -
 Microscopia de super-resolución: microscopia de colocalización (2CLM) con proteínas de fusión GFP e RFP (núcleo dunha célula de cancro óseo). 120.000 moléculas localizadas nunha área de campo ancho (470 µm2) (medido cun microscopio Vertico-SMI/SPDMphymod).
Microscopia de super-resolución: microscopia de colocalización (2CLM) con proteínas de fusión GFP e RFP (núcleo dunha célula de cancro óseo). 120.000 moléculas localizadas nunha área de campo ancho (470 µm2) (medido cun microscopio Vertico-SMI/SPDMphymod). -
 Microscopia de fluorescencia da expresión de ADN no tipo salvaxe humano e no mutante P239S da paladina.
Microscopia de fluorescencia da expresión de ADN no tipo salvaxe humano e no mutante P239S da paladina. -
 Imaxes de microscopia de fluorescencia da patoloxía de labaradas solares nunha célula sanguínea, mostrando as áreas afectadas en vermello.
Imaxes de microscopia de fluorescencia da patoloxía de labaradas solares nunha célula sanguínea, mostrando as áreas afectadas en vermello.










Notas
editar- ↑ 1,0 1,1 1,2 1,3 Spring KR, Davidson MW. "Introduction to Fluorescence Microscopy". Nikon MicroscopyU. Consultado o 2008-09-28.
- ↑ "The Fluorescence Microscope". Microscopes—Help Scientists Explore Hidden Worlds. The Nobel Foundation. Consultado o 2008-09-28.
- ↑ Juan Carlos Stockert, Alfonso Blázquez-Castro (2017). Fluorescence Microscopy in Life Sciences. Bentham Science Publishers. ISBN 978-1-68108-519-7. Arquivado dende o orixinal o 14 de maio de 2019. Consultado o 17 de decembro de 2017.
- ↑ Huang B (marzo de 2010). "Super resolution fluorescence microscopy". Annual Review of Biochemistry 78: 993–1016. PMC 2835776. PMID 19489737. doi:10.1146/annurev.biochem.77.061906.092014.
- ↑ F.A.W. Coumans; E. van der Pol; L.W.M.M. Terstappen (2012). "Flat-top illumination profile in an epi-fluorescence microscope by dual micro lens arrays". Cytometry Part A 81 (4): 324–331. PMID 22392641. doi:10.1002/cyto.a.22029.
- ↑ Colin, S., Coelho, L.P., Sunagawa, S., Bowler, C., Karsenti, E., Bork, P., Pepperkok, R. and De Vargas, C. (2017) "Quantitative 3D-imaging for cell biology and ecology of environmental microbial eukaryotes". eLife, 6: e26066. doi 10.7554/eLife.26066.002.
 O material copiouse desta fonte, que está dispoñible baixo unha Licenza Creative Commons Attribution 4.0 International.
O material copiouse desta fonte, que está dispoñible baixo unha Licenza Creative Commons Attribution 4.0 International.
- ↑ Bidhendi, AJ; Chebli, Y; Geitmann, A (maio de 2020). "Fluorescence Visualization of Cellulose and Pectin in the Primary Plant Cell Wall". Journal of Microscopy 278 (3): 164–181. PMID 32270489. doi:10.1111/jmi.12895.
- ↑ Kandel, Mikhail E.; He, Yuchen R.; Lee, Young Jae; Chen, Taylor Hsuan-Yu; Sullivan, Kathryn Michele; Aydin, Onur; Saif, M. Taher A.; Kong, Hyunjoon; Sobh, Nahil; Popescu, Gabriel (2020). "Phase imaging with computational specificity (PICS) for measuring dry mass changes in sub-cellular compartments". Nature Communications 11 (1): 6256. Bibcode:2020NatCo..11.6256K. PMC 7721808. PMID 33288761. arXiv:2002.08361. doi:10.1038/s41467-020-20062-x.
- ↑ Cremer, C; Cremer, T (1978). "Considerations on a laser-scanning-microscope with high resolution and depth of field" (PDF). Microscopica Acta 81 (1): 31–44. PMID 713859. Arquivado dende o orixinal (PDF) o 04 de marzo de 2016. Consultado o 22 de marzo de 2023.
- ↑ S.W. Hell, E.H.K. Stelzer, S. Lindek, C. Cremer; Stelzer; Lindek; Cremer (1994). "Confocal microscopy with an increased detection aperture: type-B 4Pi confocal microscopy". Optics Letters 19 (3): 222–224. Bibcode:1994OptL...19..222H. PMID 19829598. doi:10.1364/OL.19.000222.
- ↑ Baarle, Kaitlin van. "Correlative microscopy: Opening up worlds of information with fluorescence". Consultado o 2017-02-16.
- ↑ Hausmann, Michael; Schneider, Bernhard; Bradl, Joachim; Cremer, Christoph G. (1997). "High-precision distance microscopy of 3D nanostructures by a spatially modulated excitation fluorescence microscope" (PDF). En Bigio, Irving J; Schneckenburger, Herbert; Slavik, Jan; et al. Optical Biopsies and Microscopic Techniques II. Optical Biopsies and Microscopic Techniques II 3197. p. 217. doi:10.1117/12.297969. Arquivado dende o orixinal (PDF) o 04 de marzo de 2016. Consultado o 22 de marzo de 2023.
- ↑ Reymann, J; Baddeley, D; Gunkel, M; Lemmer, P; Stadter, W; Jegou, T; Rippe, K; Cremer, C; Birk, U (2008). "High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy" (PDF). Chromosome Research 16 (3): 367–82. PMID 18461478. doi:10.1007/s10577-008-1238-2. Arquivado dende o orixinal (PDF) o 04 de marzo de 2016. Consultado o 22 de marzo de 2023.
- ↑ Baddeley, David; Batram, Claudia; Weiland, Yanina; Cremer, Christoph; Birk, Udo J (2007-10). "Nanostructure analysis using spatially modulated illumination microscopy". Nature Protocols (en inglés) 2 (10): 2640–2646. ISSN 1754-2189. doi:10.1038/nprot.2007.399.
- ↑ Gunkel, M; Erdel, F; Rippe, K; Lemmer, P; Kaufmann, R; Hörmann, C; Amberger, R; Cremer, C (2009). "Dual color localization microscopy of cellular nanostructures" (PDF). Biotechnology Journal 4 (6): 927–38. PMID 19548231. doi:10.1002/biot.200900005. Arquivado dende o orixinal (PDF) o 04 de marzo de 2016. Consultado o 22 de marzo de 2023.
Véxase tamén
editarOutros artigos
editar- Imaxes e fluorescencia
- Fluorescencia nas ciencias da vida
- Microsocopia electrónica-óptica correlativa
- Elizabeth Harry, pioneira das técnicas de microscopia fluorescente para a visualización de proteínas subcelulares bacterianas
- Proteína fluorescente verde (GFP)
- Microscopio
- Desprazamento de Stokes
Ligazóns externas
editar| Wikimedia Commons ten máis contidos multimedia na categoría: Microscopio de fluorescencia |
- Microscopy Resource Center Arquivado 22 de outubro de 2014 en Wayback Machine.
- animacións e explicacións sobre varios tipos de microscopios, incluídos os microscopios fluorescente e confocais (Université Paris Sud)