PMS2

| |
| PDB 1ea6 | |
| Identificadores | |
| Símbolo | PMS2 |
| Símbolos alt. | HNPCC4, PMS2CL, PMSL2, MLH4, PMS1 homolog 2, mismatch repair system component |
| Entrez | 5395 |
| RefSeq | NP_000526 |
| UniProt | P54278 |
| Outros datos | |
| Locus | Cr. 7 7p22.1(5.97 – 6.01 Mb) |
A proteína PMS2 ou endonuclease de reparación de discordancias PMS2 é un encima que nos humanos está codificado no xene PMS2 do cromosoma 7. Intervén na reparación do ADN.[1]
Función
editarEste xene forma parte da familia de xenes PMS2, que se encontran agrupados no cromosoma 7. Os xenes relacionados con PMS2 humanos están localizados nas bandas 7p12, 7p13, 7q11 e 7q22. Os exóns do 1 ao 5 destes homólogos comparten un alto grao de identidade co PMS2 humano.[2] O produto deste xenes está implicado na reparación de discordancias do ADN. A proteína PMS2 forma un heterodímero con MLH1 e este complexo interacciona co MSH2 unido ás bases mal apareadas. Os defectos neste xenes están asociados co cancro colorrectal non poliposo hereditario, coa síndrome de Turcot e son unha causa de tumores neuroectodérmicos primitivos supratentoriais. Detectáronse variantes de empalme.[3]
Reparación de discordancias e actividade de endonuclease
editarA PMS2 intervén na reparación de discordancias no ADN e sábese que ten unha actividade latente de endonuclease que depende da integridade do motivo de meta-unión en homólogos MutL. Como endonuclease, PMS2 crea amosegas en febras de ADN descontinuas.[4]
Interaccións
editarPMS2 presenta interaccións coa proteína MLH1 ao formar con ela o heterodímero MutLα.[5][6][7][8][9][10] Hai unha competencia entre MLH3, PMS1 e PMS2 polo dominio interaccionante de MLH1, que está localizado nos residuos 492-742.[6]
Os dominios interaccionantes de PMS2 teñen repeticións de héptada que son características das proteínas con cremalleira de leucina.[6] MLH1 interacciona con PMS2 nos residuos 506-756.[7]
Os heterodímeros MutS, que son MutSα e MutSβ, asócianse con MutLα ao unirse á discordancia de bases. Crese que MutLα se une ao paso de recoñecemento de discordancias doutros procesos, como son: eliminación de discordancias da febra nova de ADN, resíntese de ADN degradado, e reparación da amosega no ADN.[10] MutLα ten unha feble actividade de ATPase e tamén posúe actividade de endonuclease que introduce amosegas na febra descontinua do ADN. Isto facilita a degradación de 5' a 3' da febra de ADN mal apareada por parte de EXO1.[10] O sitio activo de MutLα está localizado na subunidade PMS2. PMS1 e PMS2 compiten por interaccionar con MLH1.[10] As proteínas do interactoma de PMS2 foron identificadas por purificación por afinidade en tándem.[10][11]
A PMS2 humana exprésase a moi baixo nivel e non se pensa que sexa fortemente regulada no ciclo celular.[12]
Interaccións con p53 e p73
editarPMS2 tamén interacciona con p53 e p73. En ausencia de p53, as células deficientes en PMS2 e as proficientes en PMS2 aínda poden deter o ciclo celular no punto de control G2/M cando son tratadas con cisplatino.[13] As células que son deficientes en p53 e PMS2, mostran un incremento da sensibilidade a axentes anticanceríxenos. PMS2 é un mediador protector da supervivencia celular en células deficientes en p53 e modula as vías de resposta a danos no ADN protectoras independentemente de p53.[13] PMS2 e MLH1 poden protexer as células da morte celular ao contrarrestaren a apoptose mediada por p73 de maneira dependente da reparación de discordancias.[13]
PMS2 pode interaccionar con p73 para potenciar a apoptose inducida polo cisplatino ao estabilizar p73. O cisplatino estimula a interacción entre PMS2 e p73, que depende de c-Abl.[9] O complexo MutLα pode funcionar como un adaptador para traer p73 ao sitio do ADN danado e tamén actúa como activador de p73, debido á presenza de PMS2.[9] É tamén posible que o PMS2 sobreexpresado estimule a apoptose en ausencia de MLH1 e en presenza de p73 e cisplatino debido ás accións estabilizantes de PMS2 sobre p73.[9] Nos danos ao ADN, p53 induce a detención do ciclo celular por medio da vía p21/WAF e inicia a reparación por expresión de MLH1 e PMS2.[8] O complexo MSH1/PMS2 actúa como un sensor da medida dos danos no ADN, e inicia a apoptose ao estabilizar p73 se o dano non se pode reparar.[8] A perda de PMS2 non sempre conduce á inestabilidade de MLH1, xa que pode tamén formar complexos con MLH3 e PMS1.[14]
Importancia clínica
editarMutacións
editarPMS2 é un xene que codifica proteínas de reparación do ADN implicadas na reparación de discordancias. O xene PMS2 está localizado na banda 7p22 do cromosoma 7 e consta de 15 exóns. O exón 11 do xene PMS2 ten unha repetición codificada de oito adenosinas.[15]
As mutacións na liña xerminal heterocigotas en xenes de reparación de discordancias no ADN como PMS2 orixinan a síndrome de Lynch autosómica dominante. Só o 2% das familias que teñen síndrome de Lynch teñen mutacións no xene PMS2.[16] A idade dos pacientes cando se lles manifesta a síndrome de Lynch asociada a PMS2 varía moito, e vai dos 23 aos 77 anos.[17]
En raros casos, a causa desta síndrome pode ser un defecto homocigótico. En tales casos un neno herda a mutación xénica de ambos os proxenitores e a condición chámase síndrome de Turcot ou deficiencia en MMR constitucional (CMMR-D).[18] Ata o ano 2011, informouse de 36 pacientes con tumores cerebrais debidos a mutacións na liña xerminal de PMS2 bialélicas.[18] A herdanza da síndrome de Turcot pode ser dominante ou recesiva. A herdanza recesiva da síndrome de Turcot está causada por mutacións heterocigotas compostas en PMS2.[19] 31 dun total de 57 familias con CMMR-D tiñan mutacións de PMS2 na liña xerminal.[20] 19 dun total de 60 casos de portadores de mutacións en PMS2 homocigotas ou heterocigotas compostas tiñan cancro gastrointestinal ou adenomas como primeira manifestación de CMMR-D.[20] A presenza de pseudoxenes pode causar confusión cando se trata de identificar mutacións en PMS2, o que pode facer chegar á conclusión de falsos positivos sobre a presenza de PMS2 mutado.[15]
Deficiencia e sobreexpresión
editarA sobreexpresión de PMS2 ten como resultado hipermutabilidade e tolerancia aos danos no ADN.[21] A deficiencia en PMS2 tamén contribúe á inestabilidade xenética ao permitir que a mutación se propague debido á función de MMR reducida.[21] Os ratos PMS2-/- desenvolven linfomas e sarcomas. Os ratos machos PMS2-/- son estériles, o que indica que PMS2 pode ter un papel na espermatoxénese.[4]
Papel no colon normal
editar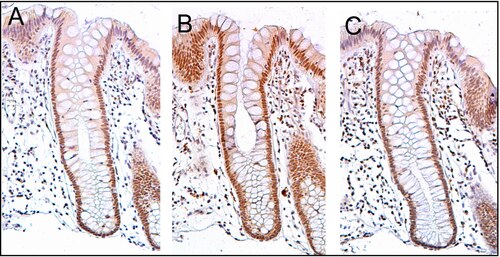
PMS2 xeralmente exprésase a alto nivel nos núcleos celulares dos enterocitos (células absorbentes) das criptas do colon que tapizan a superficie interna do colon (ver imaxe, panel A). A reparación do ADN, que implica unha alta expresión das proteínas PMS2, ERCC1 e ERCC4 (XPF), parece ser moi activa nas criptas do colon no epitelio de colon normal, non neoplástico. No caso de PMS2, o nivel de expresión no epitelio de colon normal é alto no 77% a 100% das criptas.[22]
As células orixínanse na base das criptas e migran cara a arriba ao longo do eixe da cripta antes de acabar desprendéndose no lume do colon días despois.[23] Hai 5 ou 6 células nai na base das criptas.[23] Hai uns 10 millóns de criptas ao longo da superficie interna dun colon humano medio.[22] Se as células nai na base da cripta expresan ERCC4 (XPF), xeralmente todos os miles de células da cripta expresan tamén ERCC4 (XPF). Isto vén indicado pola cor marrón que se ve por inmunomarcaxe de ERCC4 (XPF) en case todas as células da cripta no panel C da imaxe deste corte. Unha expresión similar de PMS2 e ERCC1 ocorre nas miles de células de cada cripta de colon normal.
O corte histolóxico da imaxe que se mostra aquí foi tamén contratinguido con hematoxilina para que se tinga o ADN dos núcleos de cor gris azulado. Os núcleos das células na lámina propia, que son células que están debaixo e rodeadas de criptas epiteliais, mostran intensamente a cor gris azulada que lle dá a hematoxilina e presentan pouca expresión de PMS2, ERCC1 ou ERCC4 (XPF). Ademais, as células na parte máis superior das criptas tinguidas para PMS2 (panel A) ou ERCC4 (XPF) (panel C) teñen baixos niveis destas proteínas reparadoras do ADN, polo que estas células mostran tamén a tinguidura gris azulada do ADN.[22]
Cancro de colon
editarUn 88% das células de orixe epitelial en cancros de colon, e un 50% das criptas de colon do epitelio a unha distancia de 10 cm arredor dos cancros (nos defectos de campo a partir dos cales probablemente se orixinan os cancros) teñen unha expresión de PMS2 reducida ou ausente.[22]
As deficiencias en PMS2 no epitelio do colon parecen deberse principalmente a represión epixenética. En tumores clasificados como deficientes ou carentes na reparación de discordancias, na súa maioría teñen unha expresión de PMS2 deficiente debido á falta da proteína coa que se asocia, MLH1.[24] Pairing of PMS2 with MLH1 stabilizes.[25] Comprobouse que a perda de MLH1 en cancros esporádicos debíase a silenciamento epixenetico causado por metilación do promotor en 65 de 66 casos. En 16 cancros Pms2 era deficiente incluso cando había expresión de MLH1. Destes 16 casos, non se determinou ningunha causa para 10 deles, pero en 6 atopouse unha mutación na liña xerminal heterocigota en Pms2, seguida da probable perda de heterocigose no tumor. Así, só 6 dun total de 119 tumores que carecían de expresión de Pms2 (5%) eran debidos a unha mutación en PMS2.
Coordinación con ERCC1 e ERCC4 (XPF)
editar
Cando PMS2 está reducida nas criptas do colon nun defecto de campo, isto está xeralmente asociado tamén cunha expresión reducida dos encimas de reparación do ADN ERCC1 e ERCC4 (XPF) (ver imaxes desta sección). Unha deficiencia en ERCC1 ou ERCC4 (XPF) causa unha acumulación de danos no ADN. Ese exceso de danos adoita conducir á apoptose.[26] Porén, un defectoengadido en PMS2 pode inhibir esta apoptose.[27][28] Así, unha deficiencia engadida en PMS2 probablemente sería seleccionada favorablemente nunha situación de incrementos de danos no ADN cando ERCC1 ou ERCC4 (XPF) son deficientes. Cando as células de ovario de hámster chinés deficientes para ERCC1 foron sometidas repetidamente a danos no ADN, de cinco clons derivados das células superviventes, tres tiñan mutacións en Pms2.[29]
Progresión ao cancro de colon
editarAs células de ovario de hámster chinés dobres mutantes para ERCC1 e PMS2, cando son expostas a luz ultravioleta (un axente que causa danos no ADN), mostran unha frecuencia de mutación multiplicada por 7375 en comparación co tipo silvestre, e multiplicada por 967 en comparación con células só defectivas para ERCC1.[29] Así, a deficiencia das células de colon para ERCC1 e PMS2 causa inestabilidade xenómica. Unha situación xeneticamente inestable similar espérase en células dobremente defectivas para PMS2 e ERCC4 (XPF). Esta inestabilidade probablemente potencia a progresión ao cancro de colon ao causar un fenotipo mutante,[30] e explica a presenza de células con dobre deficiencia en PMS2 e ERCC1 (ou PMS2 e ERCC4 (XPF)) en defectos de campo asociados co cancro de colon. Como indicaron Harper e Elledge,[31] os defectos na capacidade de responder axeitadamente e reparar os danos no ADN subxacen en moitas formas de cancro.
Notas
editar- ↑ Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM (Sep 1994). "Mutations of two PMS homologues in hereditary nonpolyposis colon cancer". Nature 371 (6492): 75–80. PMID 8072530. doi:10.1038/371075a0.
- ↑ Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, Vogelstein B, Kinzler KW (November 1995). "Genomic organization of the human PMS2 gene family". Genomics 30 (2): 195–206. PMID 8586419. doi:10.1006/geno.1995.9885.
- ↑ "Entrez Gene: PMS2 PMS2 postmeiotic segregation increased 2 (S. cerevisiae)".
- ↑ 4,0 4,1 van Oers JM, Roa S, Werling U, Liu Y, Genschel J, Hou H, Sellers RS, Modrich P, Scharff MD, Edelmann W (12 July 2010). "PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance". Proc. Natl. Acad. Sci. U.S.A. 107 (30): 13384–9. PMC 2922181. PMID 20624957. doi:10.1073/pnas.1008589107.
- ↑ Mac Partlin M, Homer E, Robinson H, McCormick CJ, Crouch DH, Durant ST, Matheson EC, Hall AG, Gillespie DA, Brown R (February 2003). "Interactions of the DNA mismatch repair proteins MLH1 and MSH2 with c-MYC and MAX". Oncogene 22 (6): 819–25. PMID 12584560. doi:10.1038/sj.onc.1206252.
- ↑ 6,0 6,1 6,2 Kondo E, Horii A, Fukushige S (April 2001). "The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2". Nucleic Acids Res. 29 (8): 1695–702. PMC 31313. PMID 11292842. doi:10.1093/nar/29.8.1695.
- ↑ 7,0 7,1 Guerrette S, Acharya S, Fishel R (March 1999). "The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer". J. Biol. Chem. 274 (10): 6336–41. PMID 10037723. doi:10.1074/jbc.274.10.6336.
- ↑ 8,0 8,1 8,2 Chen J, Sadowski I (March 2005). "Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements". Proc. Natl. Acad. Sci. U.S.A. 102 (13): 4813–8. PMC 555698. PMID 15781865. doi:10.1073/pnas.0407069102.
- ↑ 9,0 9,1 9,2 9,3 Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY (March 2003). "Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin". Proc. Natl. Acad. Sci. U.S.A. 100 (5): 2420–5. PMC 151356. PMID 12601175. doi:10.1073/pnas.0438031100.
- ↑ "PMS2 Gene". The GeneCards Human Gene Database. Weizmann Institute of Science.
- ↑ Meyers M, Theodosiou M, Acharya S, Odegaard E, Wilson T, Lewis JE, Davis TW, Wilson-Van Patten C, Fishel R, Boothman DA (January 1997). "Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2". Cancer Res. 57 (2): 206–8. PMID 9000555.
- ↑ 13,0 13,1 13,2 Fedier A, Ruefenacht UB, Schwarz VA, Haller U, Fink D (October 2002). "Increased sensitivity of p53-deficient cells to anticancer agents due to loss of Pms2". Br. J. Cancer 87 (9): 1027–33. PMC 2364320. PMID 12434296. doi:10.1038/sj.bjc.6600599.
- ↑ Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, Thibodeau SN, de la Chapelle A (July 2004). "Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation". Cancer Res. 64 (14): 4721–7. PMID 15256438. doi:10.1158/0008-5472.CAN-03-2879.
- ↑ 15,0 15,1 Chadwick RB, Meek JE, Prior TW, Peltomaki P, de La Chapelle A (December 2000). "Polymorphisms in a pseudogene highly homologous to PMS2". Hum. Mutat. 16 (6): 530. PMID 11102987. doi:10.1002/1098-1004(200012)16:6<530::AID-HUMU15>3.0.CO;2-6.
- ↑ "PMS2 - PMS2 postmeiotic segregation increased 2 (S. cerevisiae)". Genetics Home Reference. U.S. National Library of Medicine.
- ↑ Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A (August 2008). "The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations". Gastroenterology 135 (2): 419–28. PMC 2759321. PMID 18602922. doi:10.1053/j.gastro.2008.04.026.
- ↑ 18,0 18,1 Johannesma PC, van der Klift HM, van Grieken NC, Troost D, Te Riele H, Jacobs MA, Postma TJ, Heideman DA, Tops CM, Wijnen JT, Menko FH (September 2011). "Childhood brain tumours due to germline bi-allelic mismatch repair gene mutations". Clin. Genet. 80 (3): 243–55. PMID 21261604. doi:10.1111/j.1399-0004.2011.01635.x.
- ↑ De Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, Iannelli A, Ciciliano F, Izzo P (March 2000). "Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene". Oncogene 19 (13): 1719–23. PMID 10763829. doi:10.1038/sj.onc.1203447.
- ↑ 20,0 20,1 Herkert JC, Niessen RC, Olderode-Berends MJ, Veenstra-Knol HE, Vos YJ, van der Klift HM, Scheenstra R, Tops CM, Karrenbeld A, Peters FT, Hofstra RM, Kleibeuker JH, Sijmons RH (May 2011). "Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: case series, review and follow-up guidelines". Eur. J. Cancer 47 (7): 965–82. PMID 21376568. doi:10.1016/j.ejca.2011.01.013.
- ↑ 21,0 21,1 Gibson SL, Narayanan L, Hegan DC, Buermeyer AB, Liskay RM, Glazer PM (December 2006). "Overexpression of the DNA mismatch repair factor, PMS2, confers hypermutability and DNA damage tolerance". Cancer Lett. 244 (2): 195–202. PMID 16426742. doi:10.1016/j.canlet.2005.12.009.
- ↑ 22,0 22,1 22,2 22,3 22,4 22,5 22,6 Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr 3 (1): 3. PMC 3351028. PMID 22494821. doi:10.1186/2041-9414-3-3.
- ↑ 23,0 23,1 Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA (2014). "Quantification of crypt and stem cell evolution in the normal and neoplastic human colon". Cell Rep 8 (4): 940–7. PMC 4471679. PMID 25127143. doi:10.1016/j.celrep.2014.07.019.
- ↑ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, Cattaruzza MS, Heinimann K, Schär P, Jiricny J, Marra G (2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology 128 (5): 1160–71. PMID 15887099. doi:10.1053/j.gastro.2005.01.056.
- ↑ Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR (2000). "Steady-state regulation of the human DNA mismatch repair system". J. Biol. Chem. 275 (24): 18424–31. PMID 10747992. doi:10.1074/jbc.M001140200.
- ↑ Norbury CJ, Zhivotovsky B (2004). "DNA damage-induced apoptosis". Oncogene 23 (16): 2797–808. PMID 15077143. doi:10.1038/sj.onc.1207532.
- ↑ Fukuhara S, Chang I, Mitsui Y, Chiyomaru T, Yamamura S, Majid S, Saini S, Deng G, Gill A, Wong DK, Shiina H, Nonomura N, Lau YF, Dahiya R, Tanaka Y (2015). "Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells". Oncotarget 6 (18): 16341–51. PMC 4599273. PMID 26036629. doi:10.18632/oncotarget.3854.
- ↑ Marinovic-Terzic I, Yoshioka-Yamashita A, Shimodaira H, Avdievich E, Hunton IC, Kolodner RD, Edelmann W, Wang JY (2008). "Apoptotic function of human PMS2 compromised by the nonsynonymous single-nucleotide polymorphic variant R20Q". Proc. Natl. Acad. Sci. U.S.A. 105 (37): 13993–8. PMC 2528866. PMID 18768816. doi:10.1073/pnas.0806435105.
- ↑ 29,0 29,1 Nara K, Nagashima F, Yasui A (2001). "Highly elevated ultraviolet-induced mutation frequency in isolated Chinese hamster cell lines defective in nucleotide excision repair and mismatch repair proteins". Cancer Res. 61 (1): 50–2. PMID 11196196.
- ↑ Loeb LA (2011). "Human cancers express mutator phenotypes: origin, consequences and targeting". Nat. Rev. Cancer 11 (6): 450–7. PMC 4007007. PMID 21593786. doi:10.1038/nrc3063.
- ↑ Harper JW, Elledge SJ (2007). "The DNA damage response: ten years after". Mol. Cell 28 (5): 739–45. PMID 18082599. doi:10.1016/j.molcel.2007.11.015.
Véxase tamén
editarLigazóns externas
editar- FAQs on HNPCCArquivado 15 de agosto de 2007 en Wayback Machine. from the National Institute of Health
- GeneReviews/NCBI/NIH/UW entry on Lynch syndrome