Química bioortogonal
O termo química bioortogonal refírese a calquera reacción química que pode ocorrer dentro dos sistemas vivos sen interferir cos procesos bioquímicos nativos.[1] [2] [3] O termo bioorthogonal chemistry foi acuñado por Carolyn R. Bertozzi en 2003.[4] [5] Desde a súa introdución, o concepto de reacción bioortogonal permitiu o estudo de biomoléculas como glicanos, proteínas,[6] e lípidos[7] en tempo real en sistemas vivos sen toxicidade celular. Desenvolvéronse varias estratexias de ligadura química que cumpren os requisitos da bioortogonalidade, incluíndo a cicloadición 1,3-dipolar entre azidas e ciclooctilos (tamén denominada química de clic libre de cobre),[8] entre nitronas e ciclooctilos,[9] oxima/hidrazone formación de hidrazona a partir de aldehídos e cetonas,[10] a ligadura de tetrazina,[11] a reacción de clic baseada en isocianuros,[12] e, máis recentemente, a ligadura de cuadriciclanos.[13]
O uso da química bioortogonal adoita realizarse en dous pasos. En primeiro lugar, modifícase un substrato celular cun grupo funcional bioortogonal (reporteiro químico) e introdúcese na célula; os substratos inclúen metabolitos, inhibidores enzimáticos, etc. O informador químico non debe alterar a estrutura do substrato drasticamente para evitar afectar á súa bioactividade. En segundo lugar, introdúcese unha sonda que contén o grupo funcional complementario para reaccionar e marcar o substrato.
Aínda que se desenvolveron reaccións bioortogonais eficaces como a química de clic sen cobre, o desenvolvemento de novas reaccións segue xerando métodos ortogonais para o etiquetado que permiten utilizar múltiples métodos de etiquetado nos mesmos biosistemas. Bertozzi foi galardoada co Premio Nobel de Química en 2022 polo seu desenvolvemento da química do clic (click chemistry) e da química bioortogonal.[14]

Etimoloxía
editarA palabra bioortogonal provén do grego bio [vivo] e orthogōnios [en ángulo recto]. Así, literalmente, unha reacción que vai perpendicular a un sistema vivo, polo que non o perturba.
Requisitos de bioortogonalidade
editarPara ser considerada bioortogonal, unha reacción debe cumprir unha serie de requisitos:
- Selectividade: a reacción debe ser selectiva entre grupos funcionais endóxenos para evitar reaccións secundarias con compostos biolóxicos.
- Inercia biolóxica: os socios reactivos e o enlace resultante non deben posuír ningún modo de reactividade capaz de perturbar a funcionalidade química nativa do organismo obxecto de estudo.
- Inercia química: o enlace covalente debe ser forte e inerte para as reaccións biolóxicas.
- Cinética:A reacción debe ser rápida, na escala de tempo dos procesos celulares (minutos) para evitar a competencia en reaccións que poidan diminuír os pequenos sinais de especies menos abundantes. As reaccións rápidas tamén ofrecen unha resposta rápida, necesaria para rastrexar con precisión os procesos dinámicos.
- Biocompatibilidade das reaccións: as reaccións deben ser non tóxicas e deben funcionar en condicións biolóxicas tendo en conta o pH, os ambientes acuosos e a temperatura. A farmacocinética é unha preocupación crecente a medida que a química bioortogonal se expande a modelos animais vivos.
- Enxeñaría accesible: o informador químico debe ser capaz de incorporarse a biomoléculas mediante algunha forma de enxeñaría metabólica ou proteica. De xeito óptimo, un dos grupos funcionais tamén é moi pequeno para que non perturbe o comportamento nativo.
Enlace de Staudinger
editarBioortogonalidade
editarA azida pode actuar como un electrófilo brando que prefire os nucleófilos brandos como os fosfanos. Isto contrasta coa maioría dos nucleófilos biolóxicos que normalmente son nucleófilos duros. A reacción prodúcese selectivamente en condicións de tolerancia á auga para producir un produto estable.
As fosfinas están completamente ausentes dos sistemas vivos e non reducen os enlaces disulfuro a pesar do potencial de redución leve. As azidas demostraron ser biocompatibles en fármacos aprobados pola FDA como a azidotimidina e a través doutros usos como enlaces cruzados. Ademais, o seu pequeno tamaño permítelles incorporarse facilmente ás biomoléculas a través das vías metabólicas celulares.
Mecanismo
editarReacción clásica de Staudinger
editar
A fosfina nucleófila ataca a azida no nitróxeno terminal electrófilo. A través dun estado de transición de catro membros, o N 2 pérdese para formar un aza-iluro. O iluro inestable hidrolízase para formar óxido de fosfina e unha amina primaria. Non obstante, esta reacción non é inmediatamente bioortogonal porque a hidrólise rompe o enlace covalente no aza-iluro.
Ligadura Staudinger
editar
A reacción foi modificada para incluír un grupo éster orto ao átomo de fósforo nun dos aneis arilo para dirixir o aza-iluro a través dunha nova vía de reactividade co fin de superar a hidrólise inmediata colocando o éster para aumentar a concentración local. O ataque nucleófilo inicial á azida é o paso limitante da velocidade. O iluro reacciona coa trampa de éster electrófilo mediante ciclación intramolecular para formar un anel de cinco membros. Este anel sofre hidrólise para formar un enlace amida estable.
Limitacións
editarOs reactivos de fosfina sofren lentamente a oxidación do aire nos sistemas vivos. Ademais, é probable que sexan metabolizados in vitro por encimas do citocromo P450.
A cinética das reaccións é lenta con constantes de velocidade de segunda orde arredor de 0,0020 M −1 •s −1 . Os intentos de aumentar as taxas de ataque nucleófilo engadindo grupos doadores de electróns ás fosfinas melloraron a cinética, pero tamén aumentaron a taxa de oxidación do aire.
A cinética deficiente require que se usen altas concentracións de fosfina, o que leva a problemas co sinal de fondo elevado nas aplicacións de imaxe. Intentáronse para combater o problema do fondo elevado mediante o desenvolvemento de reactivos de fosfina fluoroxénica baseados en fluoresceína e luciferina, pero a cinética intrínseca segue sendo unha limitación.[15]
Química de clic sen cobre
editarA química de clic sen cobre é unha reacción bioortogonal desenvolvida por primeira vez por Carolyn Bertozzi como unha variante activada dunha cicloadición de Huisgen azida alquino, baseada no traballo de Karl Barry Sharpless et al. A diferenza de CuAAC, a química de clic libre de Cu foi modificada para ser bioortogonal eliminando un catalizador de cobre citotóxico, permitindo que a reacción se produza rapidamente e sen toxicidade celular viva. En lugar de cobre, a reacción é unha cicloadición de alquinazida promovida por cepas (SPAAC). Desenvolveuse como unha alternativa máis rápida á ligadura Staudinger, coas primeiras xeracións reaccionando máis de sesenta veces máis rápido. A incrible bioortogonalidade da reacción permitiu que a reacción de clic sen cobre se aplique dentro de células cultivadas, peixe cebra vivo e ratos.

Toxicidade do cobre
editarA clásica cicloadición de azida-alquino catalizada por cobre foi unha reacción de clic extremadamente rápida e eficaz para a bioconxugación, pero non é adecuada para o seu uso en células vivas debido á toxicidade dos ións Cu(I). A toxicidade débese ao dano oxidativo das especies reactivas do osíxeno formadas polos catalizadores de cobre. Tamén se atopou que os complexos de cobre inducen cambios no metabolismo celular e son absorbidos polas células.
Houbo algún desenvolvemento de ligandos para previr o dano das biomoléculas e facilitar a eliminación en aplicacións in vitro . Non obstante, descubriuse que os diferentes ambientes de ligandos dos complexos aínda poden afectar o metabolismo e a captación, introducindo unha perturbación non desexada na función celular.[16]
Bioortogonalidade
editarO grupo azida é particularmente bioortogonal porque é extremadamente pequeno (favorable para a permeabilidade celular e evita perturbacións), metabólicamente estable e non existe naturalmente nas células e, polo tanto, non ten reaccións secundarias biolóxicas competidoras. Aínda que as azidas non son o 1,3-dipolo máis reactivo dispoñible para a reacción, son preferidas pola súa relativa falta de reaccións secundarias e pola súa estabilidade en condicións sintéticas típicas.[17] O alquino non é tan pequeno, pero aínda ten a estabilidade e a ortogonalidade necesarias para o marcado in vivo. Os ciclooctinos son tradicionalmente o cicloalquino máis común para os estudos de etiquetaxe, xa que son o anel de alquino estable máis pequeno.

A reacción procede como unha cicloadición 1,3-dipolar estándar, un tipo de desprazamento pericíclico concertado asíncrono. A natureza ambivalente do 1,3-dipolo debería facer imposible a identificación dun centro electrófilo ou nucleófilo na azida de tal xeito que a dirección do fluxo de electróns cíclico careza de sentido. [p] Porén, os cálculos demostraron que a distribución de electróns entre os nitróxenos fai que o átomo de nitróxeno máis interno teña a maior carga negativa.[18]
Rexioselectividade
editarAínda que a reacción produce unha mestura rexioisomérica de triazoles, a falta de rexioselectividade na reacción non é unha gran preocupación para a maioría das aplicacións actuais. Os requisitos máis rexioespecíficos e menos bioortogonais son mellor atendidos pola cicloadición Huisgen catalizada por cobre, especialmente tendo en conta a dificultade sintética (en comparación coa adición dun alquino terminal) de sintetizar un ciclooctilo con tensión.
Desenvolvemento de ciclooctilos
editar| Ciclooctino | Constante de velocidade de segunda orde (M −1 s −1 ) |
|---|---|
| OUT | 0,0024 |
| ALO | 0,0013 |
| MOFO | 0,0043 |
| DIFO | 0,076 |
| DIBO | 0,057 |
| BARAC | 0,96 |
| DIBAC (ADIBO) | 0,31 |
| DIMAC | 0,0030 |

OCT foi o primeiro ciclooctilo desenvolvido para a química de clics sen Cu. Aínda que os alquinos lineais non son reactivos a temperaturas fisiolóxicas, a OCT foi capaz de reaccionar facilmente coas azidas en condicións biolóxicas sen mostrar toxicidade. Non obstante, era pouco soluble en auga e a cinética apenas mellorou sobre a ligadura de Staudinger. O ALO (aryl-less octyne) foi desenvolvido para mellorar a solubilidade na auga, pero aínda tiña unha cinética pobre.
Os ciclooctinos monofluorados (MOFO) e difluorados (DIFO) creáronse para aumentar a taxa mediante a adición de substituíntes de flúor que atraen electróns na posición proparxílica. O flúor é un bo grupo de atracción de electróns en termos de accesibilidade sintética e inercia biolóxica. En particular, non pode formar un aceptor de Michael electrófilo que poida reaccionar secundariamente cos nucleófilos biolóxicos.[19] DIBO (dibenzociclooctino) foi desenvolvido como unha fusión a dous aneis de arilo, o que provocou unha tensión moi elevada e unha diminución das enerxías de distorsión. Propúxose que a substitución de biarilo aumenta a tensión do anel e proporciona a conxugación co alquino para mellorar a reactividade. Aínda que os cálculos predixiron que a substitución de monoarilo proporcionaría un equilibrio óptimo entre o choque estérico (con molécula de azida) e a cepa,[20] os produtos monoarilados demostraron ser inestables.
BARAC (biarylazacyclooctynone) seguiu coa adición dun enlace amida que engade un centro tipo sp2 para aumentar a velocidade por distorsión. A resonancia de amida contribúe a unha tensión adicional sen crear insaturación adicional que levaría a unha molécula inestable. Ademais, a adición dun heteroátomo ao anel de ciclooctino mellora tanto a solubilidade como a farmacocinética da molécula. BARAC ten unha taxa (e sensibilidade) suficientes até o punto de que non é necesario eliminar o exceso de sonda para reducir o fondo. Isto fai que sexa moi útil en situacións nas que o lavado é imposible, como nas imaxes en tempo real ou nas imaxes de animais enteiros. Aínda que BARAC é extremadamente útil, a súa baixa estabilidade require que debe almacenarse a 0 °C, protexido da luz e do osíxeno.[21]
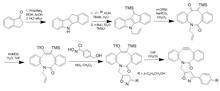
Realizáronse máis variacións de axustes en BARAC para producir DIBAC/ADIBO para engadir tensión do anel distal e reducir os estéricos ao redor do alquino para aumentar aínda máis a reactividade. O ceto-DIBO, no que o grupo hidroxilo se converteu nunha cetona, ten un aumento de tres veces na taxa debido a un cambio na conformación do anel. Os intentos de facer un difluorobenzociclooctino (DIFBO) non tiveron éxito debido á inestabilidade.
Os problemas con DIFO con estudos in vivo con ratos ilustran a dificultade de producir reaccións bioortogonais. Aínda que DIFO foi extremadamente reactivo no etiquetado das células, funcionou mal nos estudos sobre ratos debido á unión coa albumina sérica. A hidrofobicidade do ciclooctilo promove o secuestro por membranas e proteínas séricas, reducindo as concentracións biodisponibles. Como resposta, o DIMAC (dimethoxyazacyclooctyne) foi desenvolvido para aumentar a solubilidade en auga, a polaridade e a farmacocinética,[22] aínda que aínda están en desenvolvemento esforzos no etiquetado bioortogonal de modelos de rato.
Reactividade
editarOs esforzos computacionais foron vitais para explicar a termodinámica e a cinética destas reaccións de cicloadición que desempeñaron un papel vital para seguir mellorando a reacción. Existen dous métodos para activar os alquinos sen sacrificar a estabilidade: diminuír a enerxía do estado de transición ou diminuír a estabilidade dos reactivos.

Diminución da estabilidade dos reactivos: Houk[23] propuxo que as diferenzas na enerxía (Ed ‡) necesaria para distorsionar a azida e o alquino nas xeometrías do estado de transición controlan as alturas da barreira para a reacción. A enerxía de activación (E ‡) é a suma das distorsións desestabilizadoras e as interaccións estabilizadoras (Ei ‡). A distorsión máis significativa está no grupo funcional azida con menor contribución de distorsión alquino. Non obstante, é só o ciclooctino o que se pode modificar facilmente para obter unha maior reactividade. As barreiras de reacción calculadas para a azida de fenila e o acetileno (16,2 kcal/mol) fronte ao ciclooctilo (8,0 kcal/mol) dan como resultado un aumento da taxa previsto de 10 6. O ciclooctilo require menos enerxía de distorsión (1,4 kcal/mol fronte a 4,6 kcal/mol) o que resulta nunha enerxía de activación máis baixa a pesar da menor enerxía de interacción.
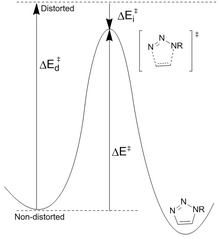
Enerxía diminuínte do estado de transición: os grupos que retiran electróns como o flúor aumentan a taxa ao diminuír a enerxía LUMO e a fenda HOMO-LUMO. Isto leva a unha maior transferencia de carga da azida ao ciclooctilo fluorado no estado de transición, aumentando a enerxía de interacción (menor valor negativo) e a enerxía de activación global.[24] A baixada do LUMO é o resultado da hiperconxugación entre os orbitais doadores alquino π e os aceptores CF σ*. Estas interaccións proporcionan estabilización principalmente no estado de transición como resultado do aumento das capacidades doadores/aceptores dos enlaces a medida que se distorsionan. Os cálculos do NBO demostraron que a distorsión do estado de transición aumenta a enerxía de interacción en 2,8 kcal/mol.
A hiperconxugación entre enlaces π fóra do plano é maior porque os enlaces π no plano están mal aliñados. Non obstante, a flexión do estado de transición permite que os enlaces π no plano teñan unha disposición máis antiperiplanar que facilita a interacción. A estabilización da enerxía da interacción hiperconxugativa adicional conséguese a través dun aumento da poboación electrónica do σ* debido á formación do enlace CN. A hiperconxugación negativa cos enlaces σ* CF mellora esta interacción estabilizadora.[25]
Rexioselectividade
editarAínda que a rexioselectividade non é un gran problema nas aplicacións actuais de imaxe da química de clic sen cobre, é un problema que impide futuras aplicacións en campos como o deseño de fármacos ou a peptidomimética.[26]
Actualmente a maioría dos ciclooctilos reaccionan para formar mesturas rexioisómeras. A análise computacional descubriu que, aínda que se calcula que a rexioselectividade da fase gaseosa favorece a adición de 1,5 sobre a adición de 1,4 até 2,9 kcal/mol en enerxía de activación, as correccións de solvatación orixinan as mesmas barreiras enerxéticas para ambos os rexioisómeros. Mentres que o isómero 1,4 na cicloadición de DIFO está desfavorecido polo seu maior momento dipolar, a solvatación estabilizao máis forte que o isómero 1,5, erosionando a rexioselectividade.[27]

Os ciclooctinos simétricos como o BCN (biciclo[6.1.0]nonino) forman un único rexioisómero tras a cicloadición[28] e poden servir para resolver este problema no futuro.
Aplicacións
editarA aplicación máis estendida da química de clic sen cobre é na imaxe biolóxica en células vivas ou animais utilizando unha biomolécula marcada con azida e un ciclooctilo que leva un axente de imaxe.
As variantes fluorescentes de ceto e oxima de DIBO utilízanse nas reaccións de clic de fluoro-interruptor nas que a fluorescencia do ciclooctilo é apagada polo triazol que se forma na reacción.[29] Por outra banda, os ciclooctinos conxugados con cumarina como coumBARAC foron desenvolvidos de xeito que o alquino suprime a fluorescencia mentres que a formación de triazol aumenta o rendemento cuántico de fluorescencia en dez veces.[30]
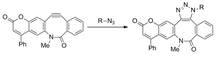
O control espacial e temporal do etiquetado do substrato investigouse mediante ciclooctilos fotoactivables. Isto permite o equilibrio do alquino antes da reacción para reducir os artefactos como resultado dos gradientes de concentración. Os ciclooctinos enmascarados son incapaces de reaccionar coas azidas na escuridade, pero convértense en alquinos reactivos ao irradiarse coa luz.[31]

Estase explorando a química de clics sen cobre para a súa utilización na síntese de axentes de imaxe PET que deben fabricarse rapidamente con gran pureza e rendemento para minimizar a desintegración isotópica antes de que se poidan administrar os compostos. Tanto as altas constantes de velocidade como a bioortogonalidade do SPAAC son susceptibles á química PET.[32]
Outras reaccións bioortogonais
editarCicloadición de dipolos de nitrón
editarA química de clic sen cobre adaptouse para usar nitronas como 1,3-dipolo en lugar de azidas e utilizouse na modificación de péptidos.[33]

Esta cicloadición entre unha nitrona e un ciclooctilo forma isoxazolinas N-alquiladas. A velocidade de reacción é mellorada pola auga e é extremadamente rápida con constantes de velocidade de segunda orde que van de 12 a 32 M −1 •s −1, dependendo da substitución da nitrona. Aínda que a reacción é extremadamente rápida, enfróntase a problemas para incorporar a nitrona ás biomoléculas mediante o etiquetado metabólico. A etiquetaxe só se conseguiu mediante a modificación do péptido postraducional.
Cicloadición de norborneno
editarAs cicloadicións dipolares 1,3 desenvolvéronse como reacción bioortogonal usando un óxido de nitrilo como 1,3-dipolo e un norborneno como dipolarófilo. O seu uso principal foi para marcar o ADN e o ARN en sintetizadores automáticos de oligonucleótidos,[34] e a reticulación de polímeros en presenza de células vivas.[35]

Os norbornenos seleccionáronse como dipolarófilos debido ao seu equilibrio entre a reactividade e a estabilidade promovidas pola cepa. Os inconvenientes desta reacción inclúen a reactividade cruzada do óxido de nitrilo debido á forte electrofilia e á cinética de reacción lenta.
Cicloadición de oxanorbornadieno
editarA cicloadición de oxanorbornadieno é unha cicloadición 1,3-dipolar seguida dunha reacción retro-Diels Alder para xerar un conxugado ligado a triazol coa eliminación dunha molécula de furano.[36] Os traballos preliminares demostraron a súa utilidade en experimentos de marcaxe peptídica e tamén se utilizou na xeración de compostos de imaxe SPECT.[37] Máis recentemente, o uso dun oxanorbornadieno foi descrito nunha reacción "iClick" a temperatura ambiente sen catalizador, na que un aminoácido modelo está ligado ao resto metálico, nunha nova aproximación ás reaccións bioortogonais.[38]

A deformación do anel e a deficiencia de electróns no oxanorbornadieno aumentan a reactividade cara ao paso que limita a taxa de cicloadición. A reacción retro-Diels Alder ocorre rapidamente despois para formar o 1,2,3 triazol estable. Os problemas inclúen unha mala tolerancia aos substituíntes que poden cambiar a electrónica do oxanorbornadieno e taxas baixas (constantes de velocidade de segunda orde da orde de 10 −4 ).
Ligadura de tetrazina
editarA ligadura de tetrazina é a reacción dun trans-cicloocteno e unha tetrazina nunha reacción de Diels Alder de demanda inversa seguida dunha reacción retro-Diels Alder para eliminar o gas nitróxeno.[39] A reacción é extremadamente rápida cunha constante de velocidade de segundo orde de 2000 M −1 –s −1 (en metanol/auga 9:1) permitindo modificacións de biomoléculas a concentracións extremadamente baixas.

Segundo o traballo computacional de Bach, a enerxía de deformación dos ciclooctenos Z é de 7,0 kcal/mol en comparación coas 12,4 kcal/mol do ciclooctano debido á perda de dúas interaccións transanulares. O E-cicloocteno ten un dobre enlace moi retorcido que dá como resultado unha enerxía de deformación de 17,9 kcal/mol.[40] Como tal, o trans-cicloocteno altamente tenso úsase como dienófilo reactivo. O dieno é unha 3,6-diaril-s-tetrazina que foi substituída para resistir a reacción inmediata coa auga. A reacción transcorre a través dunha cicloadición inicial seguida dun Diels Alder inverso para eliminar o N2 e evitar a reversibilidade da reacción.[41]
Non só a reacción tolera a auga, senón que se comprobou que a velocidade aumenta en medios acuosos. Tamén se realizaron reaccións utilizando norbornenos como dienófilos a velocidades de segunda orde da orde de 1 M −1 •s −1 en medios acuosos. A reacción aplicouse ao marcado de células vivas[42] e ao acoplamento de polímeros.[43]
[4+1] Cicloadición
editarEsta reacción de clic de isocianuro é unha cicloadición [4+1] seguida dunha eliminación retro-Diels Alder de N2.[44]

A reacción procede cunha cicloadición inicial [4+1] seguida dunha reversión para eliminar un sumidoiro termodinámico e evitar a reversibilidade. Este produto é estable se se usa unha amina terciaria ou isocianopropanoato. Se se usa un isocianuro secundario ou primario, o produto formará unha imina que se hidroliza rapidamente.
O isocianuro é un informador químico preferido debido ao seu pequeno tamaño, estabilidade, non toxicidade e ausencia nos sistemas de mamíferos. Non obstante, a reacción é lenta, con constantes de velocidade de segunda orde da orde de 10 −2 M −1 •s −1 .
Química de fotoclic de tetrazol
editarA química de Photoclick utiliza unha cicloeliminación fotoinducida para liberar N2. Isto xera un intermediario 1,3 nitrilo imina de curta duración a través da perda de gas nitróxeno, que sofre unha cicloadición 1,3-dipolar cun alqueno para xerar cicloaductos de pirazolina.[45]

A fotoindución ten lugar cunha breve exposición á luz (a lonxitude de onda depende do tetrazol) para minimizar o fotodano nas células. A reacción realízase en condicións acuosas e xera un único rexioisómero.
A nitrilo imina transitoria é moi reactiva para a cicloadición 1,3-dipolar debido a unha estrutura curvada que reduce a enerxía de distorsión. A substitución con grupos doadores de electróns nos aneis de fenilo aumenta a enerxía HOMO, cando se coloca na 1,3 nitrilo imina e aumenta a velocidade de reacción.
As vantaxes deste enfoque inclúen a capacidade de controlar espacial ou temporalmente a reacción e a capacidade de incorporar tanto alquenos como tetrazoles en biomoléculas mediante métodos biolóxicos sinxelos como a codificación xenética.[46] Ademais, o tetrazol pódese deseñar para ser fluoroxénico para controlar o progreso da reacción.[47]
Ligadura de cuadriciclano
editarA ligadura do cuadriciclano utiliza un cuadriciclano altamente tenso para someterse á cicloadición [2+2+2] con sistemas π.[48]
O cuadriciclano é abiótico, non reactivo coas biomoléculas (debido á completa saturación), relativamente pequeno e moi tenso (~80 kcal/mol). Non obstante, é moi estable a temperatura ambiente e en condicións acuosas a pH fisiolóxico. É capaz de reaccionar selectivamente con sistemas π pobres en electróns pero non con alquenos, alquinos ou cicloottinos simples.
O bis(ditiobencil)níquel(II) foi elixido como un compañeiro de reacción dunha selección de candidatos baseada na reactividade. Para evitar a reversión inducida pola luz ao norbornadieno, engádese dietilditiocarbamato para quelar o níquel do produto.
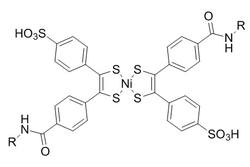
Estas reaccións son potenciadas por condicións acuosas cunha constante de velocidade de segundo orde de 0,25 M −1 •s −1 . De particular interese é que se demostrou que é bioortogonal tanto á formación de oximas como á química de clics sen cobre.
Usos
editarA química bioortogonal é unha ferramenta atractiva para a orientación previa de experimentos en imaxes nucleares e radioterapia.[49]
Notas
editar- ↑ Sletten, Ellen M.; Bertozzi, Carolyn R. (2009-09-07). "Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality". Angewandte Chemie International Edition (en inglés) 48 (38): 6974–6998. PMC 2864149. PMID 19714693. doi:10.1002/anie.200900942.
- ↑ Prescher, Jennifer A.; Dube, Danielle H.; Bertozzi, Carolyn R. (2004-08). "Chemical remodelling of cell surfaces in living animals". Nature (en inglés) 430 (7002): 873–877. ISSN 1476-4687. doi:10.1038/nature02791.
- ↑ Prescher, Jennifer A.; Dube, Danielle H.; Bertozzi, Carolyn R. (2004-08). "Chemical remodelling of cell surfaces in living animals". Nature (en inglés) 430 (7002): 873–877. ISSN 1476-4687. doi:10.1038/nature02791.
- ↑ Hang, Howard C.; Yu, Chong; Kato, Darryl L.; Bertozzi, Carolyn R. (2003-12-09). "A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation". Proceedings of the National Academy of Sciences (en inglés) 100 (25): 14846–14851. ISSN 0027-8424. PMC 299823. PMID 14657396. doi:10.1073/pnas.2335201100. Arquivado dende o orixinal o 11 de outubro de 2022. Consultado o 11 de outubro de 2022.
- ↑ Sletten, Ellen M.; Bertozzi, Carolyn R. (2011-09-20). "From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions". Accounts of Chemical Research (en inglés) 44 (9): 666–676. ISSN 0001-4842. PMC 3184615. PMID 21838330. doi:10.1021/ar200148z.
- ↑ Plass, Tilman; Milles, Sigrid; Koehler, Christine; Schultz, Carsten; Lemke, Edward A. (2011-04-18). "Genetically Encoded Copper-Free Click Chemistry". Angewandte Chemie International Edition (en inglés) 50 (17): 3878–3881. PMC 3210829. PMID 21433234. doi:10.1002/anie.201008178.
- ↑ Neef, Anne B.; Schultz, Carsten (2009-02-09). "Selective Fluorescence Labeling of Lipids in Living Cells". Angewandte Chemie International Edition (en inglés) 48 (8): 1498–1500. ISSN 1433-7851. doi:10.1002/anie.200805507.
- ↑ Baskin, Jeremy M.; Prescher, Jennifer A.; Laughlin, Scott T.; Agard, Nicholas J.; Chang, Pamela V.; Miller, Isaac A.; Lo, Anderson; Codelli, Julian A.; Bertozzi, Carolyn R. (2007-10-23). "Copper-free click chemistry for dynamic in vivo imaging". Proceedings of the National Academy of Sciences (en inglés) 104 (43): 16793–16797. ISSN 0027-8424. PMC 2040404. PMID 17942682. doi:10.1073/pnas.0707090104. Arquivado dende o orixinal o 11 de outubro de 2022. Consultado o 11 de outubro de 2022.
- ↑ Ning, Xinghai; Temming, Rinske P.; Dommerholt, Jan; Guo, Jun; Ania, Daniel B.; Debets, Marjoke F.; Wolfert, Margreet A.; Boons, Geert-Jan; van Delft, Floris L. (2010-03-23). "Protein Modification by Strain-Promoted Alkyne-Nitrone Cycloaddition". Angewandte Chemie International Edition (en inglés) 49 (17): 3065–3068. PMC 2871956. PMID 20333639. doi:10.1002/anie.201000408.
- ↑ Yarema, Kevin J.; Mahal, Lara K.; Bruehl, Richard E.; Rodriguez, Elena C.; Bertozzi, Carolyn R. (1998-11-20). "Metabolic Delivery of Ketone Groups to Sialic Acid Residues: APPLICATION TO CELL SURFACE GLYCOFORM ENGINEERING *". Journal of Biological Chemistry (en English) 273 (47): 31168–31179. ISSN 0021-9258. PMID 9813021. doi:10.1074/jbc.273.47.31168.
- ↑ Blackman, Melissa L.; Royzen, Maksim; Fox, Joseph M. (2008-10-15). "Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity". Journal of the American Chemical Society (en inglés) 130 (41): 13518–13519. ISSN 0002-7863. PMC 2653060. PMID 18798613. doi:10.1021/ja8053805.
- ↑ Stöckmann, Henning; Neves, André A.; Stairs, Shaun; Brindle, Kevin M.; Leeper, Finian J. (2011-10-12). "Exploring isonitrile-based click chemistry for ligation with biomolecules". Organic & Biomolecular Chemistry (en inglés) 9 (21): 7303–7305. ISSN 1477-0539. doi:10.1039/C1OB06424J.
- ↑ Sletten, Ellen M.; Bertozzi, Carolyn R. (2011-11-09). "A Bioorthogonal Quadricyclane Ligation". Journal of the American Chemical Society (en inglés) 133 (44): 17570–17573. ISSN 0002-7863. PMC 3206493. PMID 21962173. doi:10.1021/ja2072934.
- ↑ "Chemistry". The Nobel Prize; nobelprize.org.
- ↑ Chang, Pamela V.; Prescher, Jennifer A.; Hangauer, Matthew J.; Bertozzi, Carolyn R. (2007-07-01). "Imaging Cell Surface Glycans with Bioorthogonal Chemical Reporters". Journal of the American Chemical Society (en inglés) 129 (27): 8400–8401. ISSN 0002-7863. doi:10.1021/ja070238o.
- ↑ Kennedy, David C.; McKay, Craig S.; Legault, Marc C. B.; Danielson, Dana C.; Blake, Jessie A.; Pegoraro, Adrian F.; Stolow, Albert; Mester, Zoltan; Pezacki, John Paul (2011-11-09). "Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions". Journal of the American Chemical Society (en inglés) 133 (44): 17993–18001. ISSN 0002-7863. doi:10.1021/ja2083027.
- ↑ Huisgen, Rolf (1976-02). "1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates". The Journal of Organic Chemistry (en inglés) 41 (3): 403–419. ISSN 0022-3263. doi:10.1021/jo00865a001.
- ↑ Gold, Brian; Shevchenko, Nikolay E.; Bonus, Natalie; Dudley, Gregory B.; Alabugin, Igor V. (2012-01-06). "Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry". The Journal of Organic Chemistry (en inglés) 77 (1): 75–89. ISSN 0022-3263. doi:10.1021/jo201434w.
- ↑ Baskin, Jeremy M.; Prescher, Jennifer A.; Laughlin, Scott T.; Agard, Nicholas J.; Chang, Pamela V.; Miller, Isaac A.; Lo, Anderson; Codelli, Julian A.; Bertozzi, Carolyn R. (2007-10-23). "Copper-free click chemistry for dynamic in vivo imaging". Proceedings of the National Academy of Sciences (en inglés) 104 (43): 16793–16797. ISSN 0027-8424. PMC 2040404. PMID 17942682. doi:10.1073/pnas.0707090104. Arquivado dende o orixinal o 11 de outubro de 2022. Consultado o 11 de outubro de 2022.
- ↑ Chenoweth, Kimberly; Chenoweth, David; Iii, William A. Goddard (2009-11-25). "Cyclooctyne-based reagents for uncatalyzed click chemistry: A computational survey". Organic & Biomolecular Chemistry (en inglés) 7 (24): 5255–5258. ISSN 1477-0539. doi:10.1039/B911482C.
- ↑ Jewett, John C.; Sletten, Ellen M.; Bertozzi, Carolyn R. (2010-03-24). "Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones". Journal of the American Chemical Society (en inglés) 132 (11): 3688–3690. ISSN 0002-7863. PMC 2840677. PMID 20187640. doi:10.1021/ja100014q.
- ↑ Sletten, Ellen M.; Bertozzi, Carolyn R. (2008-07-17). "A Hydrophilic Azacyclooctyne for Cu-Free Click Chemistry". Organic Letters (en inglés) 10 (14): 3097–3099. ISSN 1523-7060. PMC 2664610. PMID 18549231. doi:10.1021/ol801141k.
- ↑ Ess, Daniel H.; Jones, Gavin O.; Houk, K. N. (2008-04-01). "Transition States of Strain-Promoted Metal-Free Click Chemistry: 1,3-Dipolar Cycloadditions of Phenyl Azide and Cyclooctynes". Organic Letters (en inglés) 10 (8): 1633–1636. ISSN 1523-7060. doi:10.1021/ol8003657.
- ↑ Schoenebeck, Franziska; Ess, Daniel H.; Jones, Gavin O.; Houk, K. N. (2009-06-17). "Reactivity and Regioselectivity in 1,3-Dipolar Cycloadditions of Azides to Strained Alkynes and Alkenes: A Computational Study". Journal of the American Chemical Society (en inglés) 131 (23): 8121–8133. ISSN 0002-7863. doi:10.1021/ja9003624.
- ↑ Gold, Brian; Shevchenko, Nikolay E.; Bonus, Natalie; Dudley, Gregory B.; Alabugin, Igor V. (2012-01-06). "Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry". The Journal of Organic Chemistry (en inglés) 77 (1): 75–89. ISSN 0022-3263. doi:10.1021/jo201434w.
- ↑ Lutz, Jean-François (2008-03-07). "Copper-Free Azide–Alkyne Cycloadditions: New Insights and Perspectives". Angewandte Chemie International Edition (en inglés) 47 (12): 2182–2184. doi:10.1002/anie.200705365.
- ↑ Schoenebeck, Franziska; Ess, Daniel H.; Jones, Gavin O.; Houk, K. N. (2009-06-17). "Reactivity and Regioselectivity in 1,3-Dipolar Cycloadditions of Azides to Strained Alkynes and Alkenes: A Computational Study". Journal of the American Chemical Society (en inglés) 131 (23): 8121–8133. ISSN 0002-7863. doi:10.1021/ja9003624.
- ↑ Dommerholt, Jan; Schmidt, Samuel; Temming, Rinske; Hendriks, Linda J. A.; Rutjes, Floris P. J. T.; van Hest, Jan C. M.; Lefeber, Dirk J.; Friedl, Peter; van Delft, Floris L. (2010-12-03). "Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells". Angewandte Chemie International Edition (en inglés) 49 (49): 9422–9425. PMC 3021724. PMID 20857472. doi:10.1002/anie.201003761.
- ↑ Mbua, Ngalle Eric; Guo, Jun; Wolfert, Margreet A.; Steet, Richard; Boons, Geert-Jan (2011-08-16). "Strain-Promoted Alkyne-Azide Cycloadditions (SPAAC) Reveal New Features of Glycoconjugate Biosynthesis". ChemBioChem (en inglés) 12 (12): 1912–1921. PMC 3151320. PMID 21661087. doi:10.1002/cbic.201100117.
- ↑ Jewett, John C.; Bertozzi, Carolyn R. (2011-11-18). "Synthesis of a Fluorogenic Cyclooctyne Activated by Cu-Free Click Chemistry". Organic Letters (en inglés) 13 (22): 5937–5939. ISSN 1523-7060. PMC 3219546. PMID 22029411. doi:10.1021/ol2025026.
- ↑ Poloukhtine, Andrei A.; Mbua, Ngalle Eric; Wolfert, Margreet A.; Boons, Geert-Jan; Popik, Vladimir V. (2009-11-04). "Selective Labeling of Living Cells by a Photo-Triggered Click Reaction". Journal of the American Chemical Society (en inglés) 131 (43): 15769–15776. ISSN 0002-7863. PMC 2776736. PMID 19860481. doi:10.1021/ja9054096.
- ↑ Carpenter, Richard D.; Hausner, Sven H.; Sutcliffe, Julie L. (2011-12-08). "Copper-Free Click for PET: Rapid 1,3-Dipolar Cycloadditions with a Fluorine-18 Cyclooctyne". ACS Medicinal Chemistry Letters (en inglés) 2 (12): 885–889. ISSN 1948-5875. PMC 4018166. PMID 24900276. doi:10.1021/ml200187j.
- ↑ Ning, Xinghai; Temming, Rinske P.; Dommerholt, Jan; Guo, Jun; Ania, Daniel B.; Debets, Marjoke F.; Wolfert, Margreet A.; Boons, Geert-Jan; van Delft, Floris L. (2010-03-23). "Protein Modification by Strain-Promoted Alkyne-Nitrone Cycloaddition". Angewandte Chemie International Edition (en inglés) 49 (17): 3065–3068. PMC 2871956. PMID 20333639. doi:10.1002/anie.201000408.
- ↑ Gutsmiedl, Katrin; Wirges, Christian T.; Ehmke, Veronika; Carell, Thomas (2009-06-04). "Copper-Free “Click” Modification of DNA via Nitrile Oxide−Norbornene 1,3-Dipolar Cycloaddition". Organic Letters (en inglés) 11 (11): 2405–2408. ISSN 1523-7060. doi:10.1021/ol9005322.
- ↑ Truong, Vinh X.; Zhou, Kun; Simon, George P.; Forsythe, John S. (2015-10). "Nitrile Oxide-Norbornene Cycloaddition as a Bioorthogonal Crosslinking Reaction for the Preparation of Hydrogels". Macromolecular Rapid Communications 36 (19): 1729–1734. ISSN 1521-3927. PMID 26250120. doi:10.1002/marc.201500314.
- ↑ van Berkel, Sander S.; Dirks, A. (Ton) J.; Debets, Marjoke F.; van Delft, Floris L.; Cornelissen, Jeroen J. L. M.; Nolte, Roeland J. M.; Rutjes, Floris P. J. T. (2007-09-03). "Metal-Free Triazole Formation as a Tool for Bioconjugation". ChemBioChem (en inglés) 8 (13): 1504–1508. doi:10.1002/cbic.200700278.
- ↑ van Berkel, Sander S.; Dirks, A. (Ton) J.; Meeuwissen, Silvie A.; Pingen, Dennis L. L.; Boerman, Otto C.; Laverman, Peter; van Delft, Floris L.; Cornelissen, Jeroen J. L. M.; Rutjes, Floris P. J. T. (2008-07-21). "Application of Metal‐Free Triazole Formation in the Synthesis of Cyclic RGD–DTPA Conjugates". ChemBioChem (en inglés) 9 (11): 1805–1815. doi:10.1002/cbic.200800074.
- ↑ Henry, Lucas; Schneider, Christoph; Mützel, Benedict; Simpson, Peter V.; Nagel, Christoph; Fucke, Katharina; Schatzschneider, Ulrich (2014-11-18). "Amino acid bioconjugation via iClick reaction of an oxanorbornadiene-masked alkyne with a MnI(bpy)(CO)3-coordinated azide". Chemical Communications (en inglés) 50 (99): 15692–15695. ISSN 1364-548X. doi:10.1039/C4CC07892F.
- ↑ Row, R. David; Prescher, Jennifer A. (2016-08-24). "Tetrazine Marks the Spot". ACS Central Science (en inglés) 2 (8): 493–494. ISSN 2374-7943. PMC 4999966. PMID 27610408. doi:10.1021/acscentsci.6b00204.
- ↑ Bach, Robert D. (2009-04-15). "Ring Strain Energy in the Cyclooctyl System. The Effect of Strain Energy on [3 + 2] Cycloaddition Reactions with Azides". Journal of the American Chemical Society (en inglés) 131 (14): 5233–5243. ISSN 0002-7863. doi:10.1021/ja8094137.
- ↑ Blackman, Melissa L.; Royzen, Maksim; Fox, Joseph M. (2008-10-15). "Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity". Journal of the American Chemical Society (en inglés) 130 (41): 13518–13519. ISSN 0002-7863. PMC 2653060. PMID 18798613. doi:10.1021/ja8053805.
- ↑ Devaraj, Neal K.; Weissleder, Ralph; Hilderbrand, Scott A. (2008-12-17). "Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging". Bioconjugate Chemistry (en inglés) 19 (12): 2297–2299. ISSN 1043-1802. PMC 2677645. PMID 19053305. doi:10.1021/bc8004446.
- ↑ Hansell, Claire F.; Espeel, Pieter; Stamenović, Milan M.; Barker, Ian A.; Dove, Andrew P.; Du Prez, Filip E.; O’Reilly, Rachel K. (2011-09-07). "Additive-Free Clicking for Polymer Functionalization and Coupling by Tetrazine–Norbornene Chemistry". Journal of the American Chemical Society (en inglés) 133 (35): 13828–13831. ISSN 0002-7863. doi:10.1021/ja203957h.
- ↑ Stöckmann, Henning; Neves, André A.; Stairs, Shaun; Brindle, Kevin M.; Leeper, Finian J. (2011-10-12). "Exploring isonitrile-based click chemistry for ligation with biomolecules". Organic & Biomolecular Chemistry (en inglés) 9 (21): 7303–7305. ISSN 1477-0539. doi:10.1039/C1OB06424J.
- ↑ Stöckmann, Henning; Neves, André A.; Stairs, Shaun; Brindle, Kevin M.; Leeper, Finian J. (2011-10-12). "Exploring isonitrile-based click chemistry for ligation with biomolecules". Organic & Biomolecular Chemistry (en inglés) 9 (21): 7303–7305. ISSN 1477-0539. doi:10.1039/C1OB06424J.
- ↑ Lim, Reyna K. V.; Lin, Qing (2011-09-20). "Photoinducible Bioorthogonal Chemistry: A Spatiotemporally Controllable Tool to Visualize and Perturb Proteins in Live Cells". Accounts of Chemical Research (en inglés) 44 (9): 828–839. ISSN 0001-4842. PMC 3175026. PMID 21609129. doi:10.1021/ar200021p.
- ↑ Song, Wenjiao; Wang, Yizhong; Qu, Jun; Lin, Qing (2008-07). "Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells". Journal of the American Chemical Society (en inglés) 130 (30): 9654–9655. ISSN 0002-7863. doi:10.1021/ja803598e.
- ↑ Sletten, Ellen M.; Bertozzi, Carolyn R. (2011-11-09). "A Bioorthogonal Quadricyclane Ligation". Journal of the American Chemical Society (en inglés) 133 (44): 17570–17573. ISSN 0002-7863. PMC 3206493. PMID 21962173. doi:10.1021/ja2072934.
- ↑ Knight, James C; Cornelissen, Bart (2014-03-20). "Bioorthogonal chemistry: implications for pretargeted nuclear (PET/SPECT) imaging and therapy". American Journal of Nuclear Medicine and Molecular Imaging 4 (2): 96–113. ISSN 2160-8407. PMC 3992206. PMID 24753979.